|
ホーム みなさんに 履歴書 新見正則の理念 趣味・独り言 医学生から今までの軌跡 投稿文・エッセイ 著書 TV どらえもんラボ 血管外科医募集 オックスフォード大学博士論文 英文論文 リンク |
オックスフォード大学博士論文
以下は私のオックスフォード大学での博士課程(Degree of Philosohy)取得のための論文です。
300頁におよぶ製本されたもので、原本はオックスフォード大学のボードリアンライブラリーに永久保存されています。
An investigation to determine the ability of allogeneic resting B cells to induce specific unresponsiveness in vivo by and Nuffield Department of Surgery A thesis submitted in partial fulfillment of the requirements for
Dedications To Tomoe |
Acknowledgements First, and foremost, I would like to thank Professor Kathryn Wood, in particular, for her support and encouragement, and for allowing me the opportunity to pursue my research in her laboratory to the successful completion of my D. Phil.. I would also like to thank Professor Sir Peter Morris for giving me the opportunity to work in the Nuffield Department of Surgery and for being a very special source of inspiration and motivation. In addition, I would like to thank the following: To Thomas Pearson and Christian Larsen for giving me the opportunity to carry out experiments at Emory University in Atlanta. Also, I would like to thank Alex, Meghan and Caroline Pearson for their very kind hospitality to my wife, Tomoe, and myself during our stay in Atlanta. To Andrew Mellor, Raif Geha and Elaine Thomas for providing the CBK, CD40 knockout and gp39 knockout mice, respectively. To Diane Hollenbaugh and Alejandro Aruffo for providing the anti-gp39 monoclonal antibody and to Peter Linsley for providing the CTLA4-Ig. To Professor Herman Waldmann for providing the anti-CD4 and CD8 monoclonal antibody hybridomas, YTS191 and YTS169, respectively. To Nigel Rust for his assistance with the cell sorter, Gareth Plant for his assistance with tissue culture and David Hughes for performing the Paraffin histology. To thank all members of the Wood group and members of the Department for their friendly help, fruitful discussion and for their tolerance, help and support in correcting my English, whenever needed, especially Andrew Bushell, Nick Jones, Dave Roelen, Masaki Hara, Wilson Wong, Bryant Gilot, Jeff Barbara, Stuart Turvey, Jeremy Fry and Nick Fluck, and Mary-Anne Rawsthorne who taught me the technique of skin grafting. To the staff at the Biomedical Services Unit for their expert care of the animals used in this study. To Teikyo University Japan which provided the funding to allow me to undertake a D. Phil. at the Nuffield Department of Surgery. To John Thomas and Jackie Maher for taking care of our private and official life in Oxford. To Professor Susumu Kodaira for allowing me to study transplant immunology for five years in Oxford. Finally, I would like to offer my utmost thanks and gratitude to Dr. Shoichi Okinaga, President of Teikyo University, Japan.
|
Abstract An investigation to determine the ability of allogeneic resting B cells to induce specific unresponsiveness in vivo ---------------------------------------------- Masanori Niimi D. Phil Thesis |
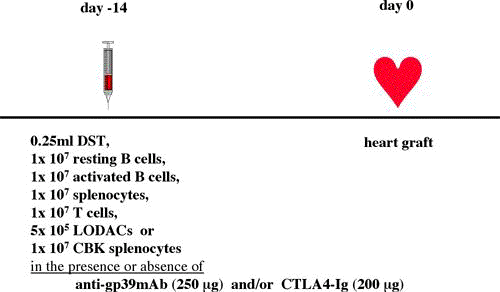
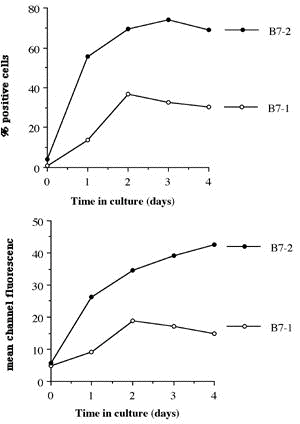
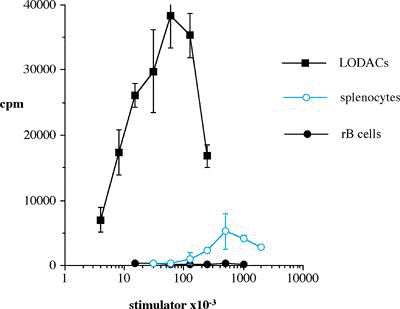
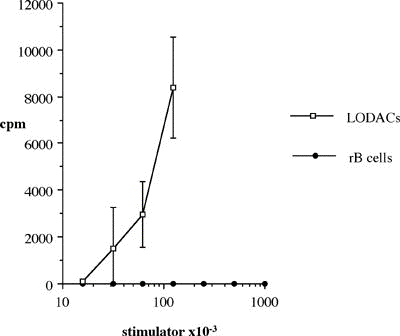
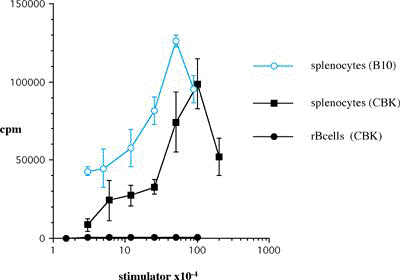
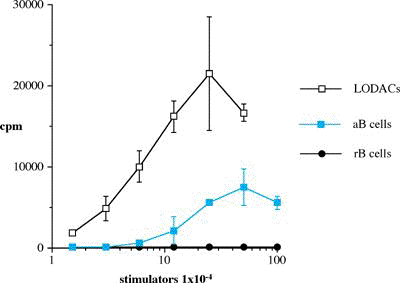
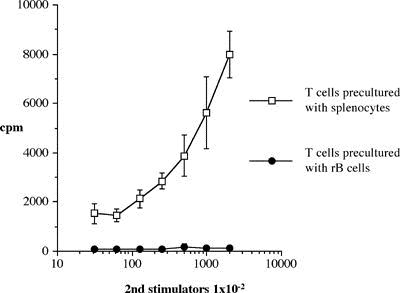
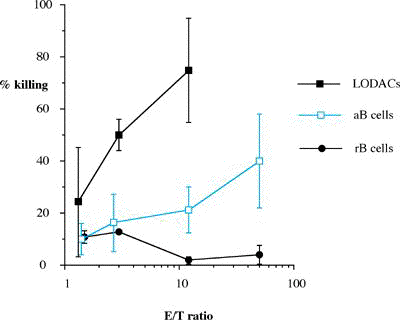
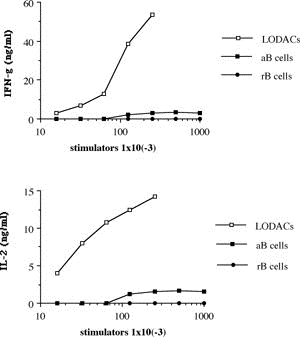
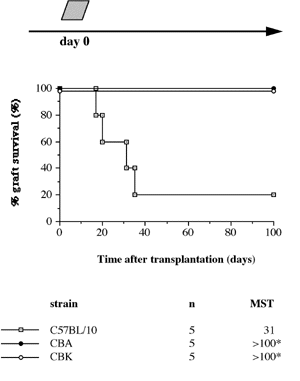
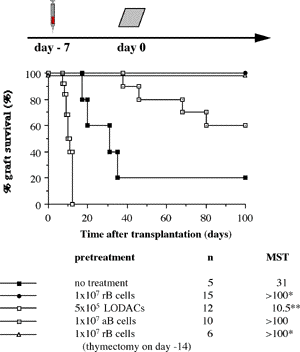
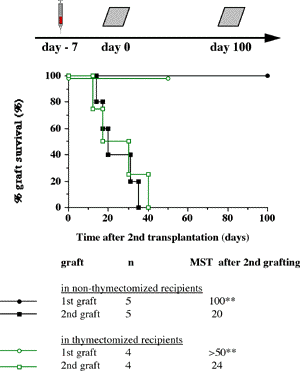
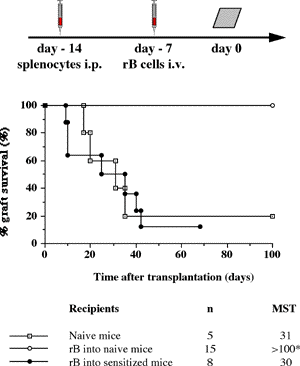
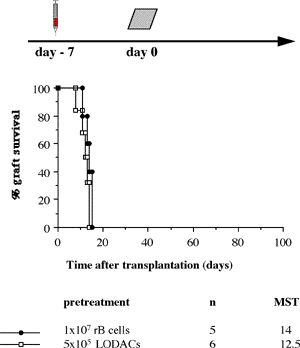
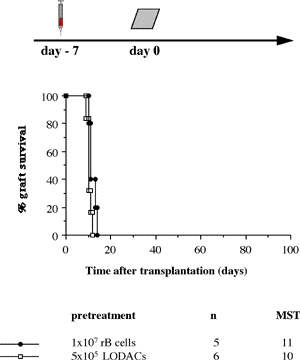
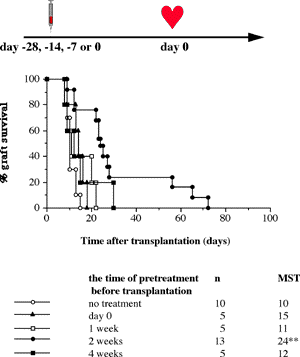
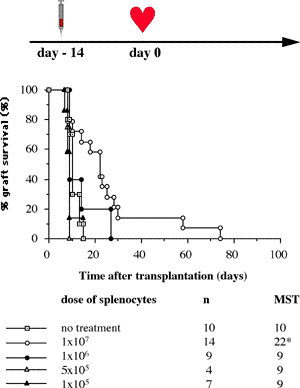
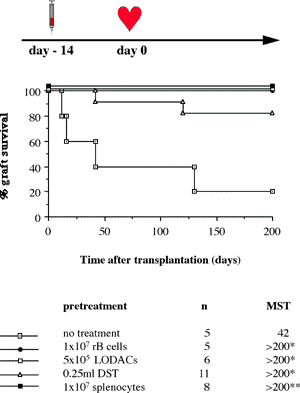
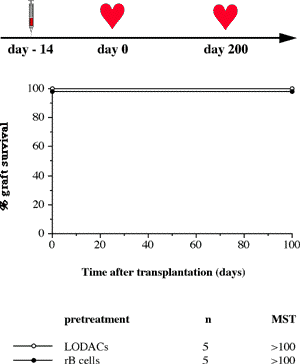
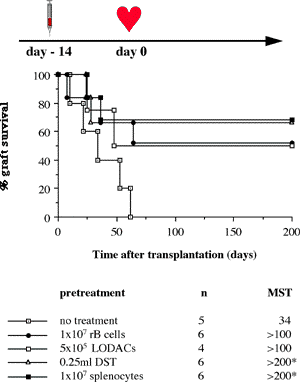
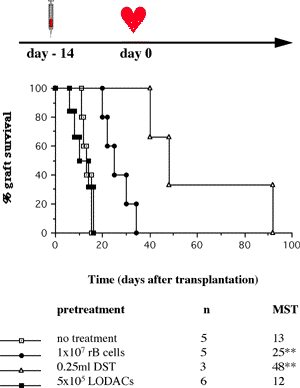
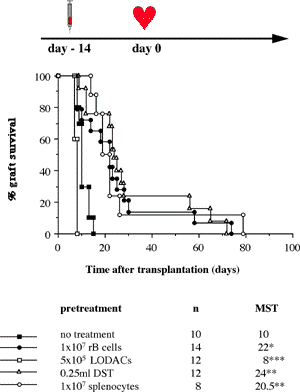
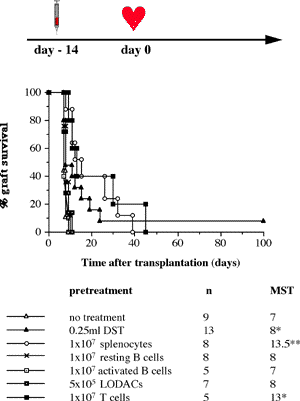
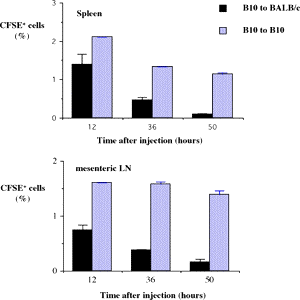
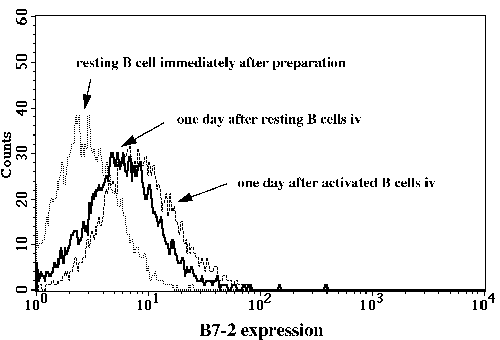
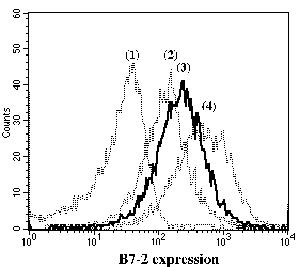
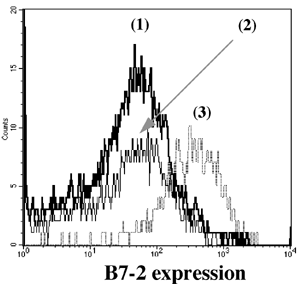
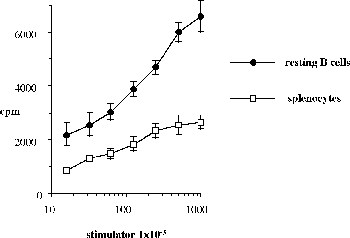
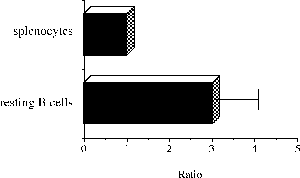
One of the final goals for transplantation immunology is to achieve and solve the mechanism of donor-specific tolerance. Donor-specific transfusions have been shown to induce donor-specific unresponsiveness. However, the mechanism responsible is still unclear. Resting B cells (rB cells) have been shown to be incompetent antigen presenting cells (APCs) in vitro. Pretreatment with rB cells in vivo has been shown to result in the induction of specific unresponsiveness to skin grafts mismatched for a single minor histocompatibility (miH) antigen, H-Y. We therefore examined the possibility that rB cells may prolong the survival of grafts mismatched for miH and/or major histocompatibility complex (MHC) antigens. First we confirmed that pretreatment with donor rB cells but not low density adherent cells (LODACs), a population enriched for dendritic cells, was able to induce prolongation of skin grafts mismatched for a single miH antigen, H-Y. In contrast, rB cells and LODACs were found to be equally effective at prolonging the survival of cardiac grafts mismatched for miH antigen(s). However, when donors and recipients were mismatched for MHC antigens, pretreatment with rB cells was ineffective in prolonging the survival of either heart or skin grafts. We hypothesised that the reason why rB cells were ineffective in this situation was due to activation of the rB cells in vivo. B7-2 expression was detected on allogeneic rB cells within 24 hours of injection in the spleens of recipient mice. There are at least two other possibilities to explain our observation. First, rB cells might turn off naive but not activated memory T cells. As alloreactive T cells can be cross-reactive with environmental antigens, a larger population of the repertoire might be antigen experienced and therefore resistant to the effect of the rB cells. Second, donor antigens on rB cells might be presented on recipient APCs, and that T cells activated by this route could induce rejection. |
Abbreviations aB cells Activated B cells |
Publications The results in this thesis have been published or submitted as follows: (1) Niimi M, Rawsthorne MA, Morris PJ , Wood KJ (2) Niimi M, Morris PJ , Wood KJ (3) Niimi M, Roelen D, Wong W, Hara M, Morris PJ, Wood KJ (4) Niimi M, Pearson TC, Larsen CP, Alexander DZ, Hollenbaugh D, Aruffo A, Linsley PS, Morris PJ, Wood KJ |
Contents |
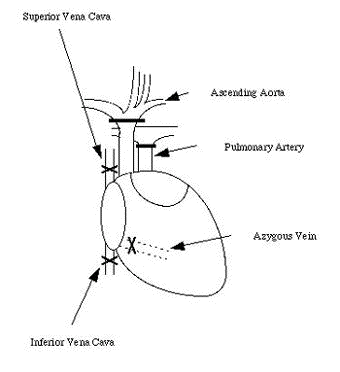
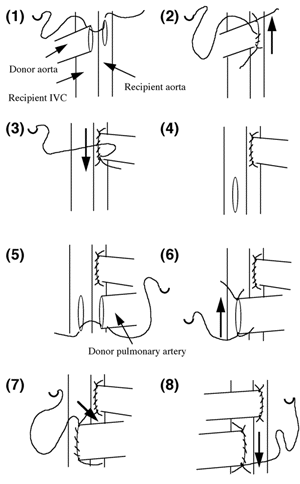
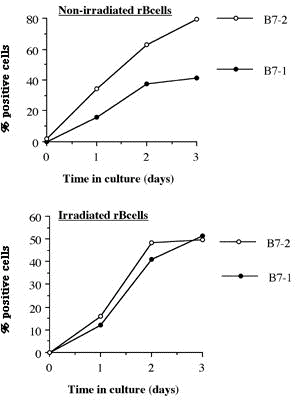
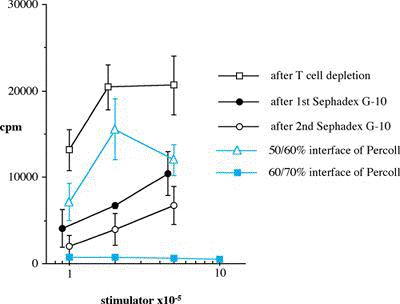
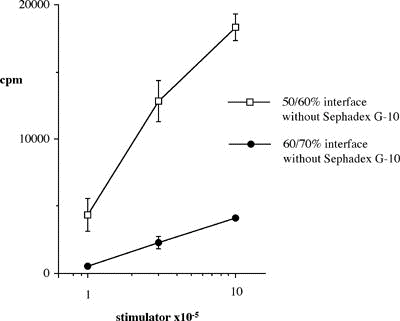
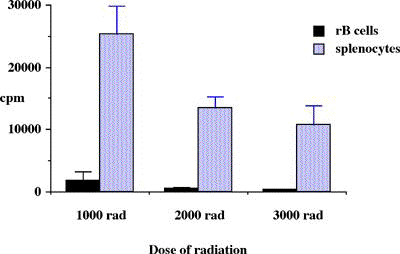
Abstract ............................................ 5 Abbreviations ....................................... 6 Publications ........................................ 8 Chapter 1: Introduction 1.1 Clinical aspects 1.1.1 Introduction..................................... 1 1.1.2 Historical development........................... 2 1.1.3 Current immunosuppression........................ 3 1.2 Graft rejection 1.2.1 Immune recognition .............................. 5 1.2.2 Immune rejection of allografts................... 5 1.2.3 Direct and indirect pathway of allorecognition... 8 1.2.4 H-Y antigen...................................... 9 1.3 Mechanisms of tolerance 1.3.1 Historical perspective of immunological tolerance......................... 11 1.3.2 Deletion of self-reactive T cells............... 12 1.3.3 The existence of self-reactive T cells in the periphery................................ 13 1.3.4 History of anergy, "functional deletion"........ 13 1.3.5 Deletion and anergy of B cells.................. 15 1.3.6 Evidence for an active mechanism of tolerance... 16 1.3.7 Infectious tolerance............................ 17 1.3.8 Immune deviation................................ 18 1.3.9 T cell ignorance................................ 19 1.4 Antigen presenting cells (APCs) 1.4.1 Historical context.............................. 20 1.4.2 Dendritic cells................................. 21 1.4.3 B cells as APCs................................. 22 1.4.4 APC in transplantation.......................... 23 1.5 Costimulatory molecules 1.5.1 B7 / CD28 pathway............................... 26 1.5.1.1 Expression of CD28 / CTLA4.................... 26 1.5.1.2 Regulation of T cell activation by CD28 and CTLA4............................. 27 1.5.1.3 B7-1 and B7-2................................. 29 1.5.1.4 Different functional effects of B7-1 and B7-2. 30 1.5.2 gp39 / CD40 pathway............................. 31 1.6 Protocols to induce operational tolerance 1.6.1 Acquired tolerance.............................. 34 1.6.2 The blood transfusion effect.................... 34 1.6.3 The use of mAbs to modulate immune responsiveness in the adult.................................... 35 1.6.3.1 Anti-CD4 mAb.................................. 36 1.6.3.2 Anti-LFA-1 mAb................................ 37 1.6.3.3 Anti-IL-2 receptor mAb........................ 37 1.6.3.4 Anti-B7 mAb and CTLA4-Ig...................... 37 1.6.3.5 Anti-gp39 mAb................................. 38 1.6.4 Tolerance induction prior to transplantation.... 39 1.6.4.1 Anti-CD4 mAb + alloantigen.................... 39 1.6.4.2 CTLA4-Ig + alloantigen........................ 40 1.6.4.3 Anti-gp39 + B cells........................... 41 1.7 General aims of the thesis........................ 43 1.8 Figures........................................... 45 Chapter 2: Materials and Methods 2.1 Animals 2.1.1 Mice............................................ 48 2.1.2 Rats............................................ 49 2.2 Tissue culture media and reagents for cell preparation Phosphate buffered saline (PBS)....................... 50 Heat inactivated Foetal Calf Serum (FCS).............. 50 L-glutamine........................................... 50 Antibiotics........................................... 50 2-mercapto-ethanol (2ME).............................. 51 RPMI1640.............................................. 51 Complete RPMI tissue culture medium (R10)............. 51 DMEM.................................................. 51 Complete DMEM......................................... 51 PBS/EDTA.............................................. 52 Tris-buffered ammonium chloride (TBAC)................ 52 Sephadex G-10......................................... 52 Percoll............................................... 52 "Dense" bovine serum albumin (BSA) fraction V......... 52 Lipopolysaccharide (LPS).............................. 53 Dimethyl sulphoxide (DMSO)............................ 53 Phorbol myristate acetate (PMA)....................... 53 Ionomycin............................................. 53 3H-thymidine.......................................... 53 Chromium 51 (51Cr).................................... 53 Con A pulsed spleen cell supernatant medium........... 54 Sodium azide (NaN3)................................... 54 Triton-X 100 solution................................. 54 Freezing medium....................................... 54 2.3 Cell line 2.3.1 RMA and P815.................................... 55 2.3.2 L-Kb cells...................................... 55 2.4 Stock cells 2.4.1 Freezing........................................ 55 2.4.2 Thawing......................................... 55 2.5 Preparation of specific cell populations 2.5.1 Preparation of splenocytes...................... 57 2.5.2 Preparation of B cells.......................... 57 2.5.3 Preparation of Low Density Adherent Cells (LODACs)........................................ 58 2.5.4 Preparation of nylon wool purified T cells...... 59 2.5.5 Preparation of FACS sorted T cells.............. 59 2.6 Flow cytometry 2.6.1 Normal staining................................. 60 2.6.2 Staining of B7-2 on IAb+ cells.................. 60 2.6.3 Staining cells for tracing in vivo.............. 61 2.7 Allo-antibody assay............................... 63 2.8 Functional assay in vitro 2.8.1 Mixed leukocyte culture (MLC)................... 64 2.8.2 Labelling 51Chromium............................ 64 2.8.3 Cytotoxic T lymphocyte (CTL) assay.............. 64 2.8.4 Limiting dilution analysis (LDA)................ 64 2.9 Enzyme-linked immunosorbent assay (ELISA) 2.9.1 Reagents for ELISA.............................. 66 2.9.2 Methods of ELISA................................ 66 2.9.3 mAbs for ELISA.................................. 68 2.10 Monoclonal antibody (mAb) purification 2.10.1 Production of mAb in tissue culture supernatant.................................... 69 2.10.2 Production of mAb from rat ascites injected with hybridoma (1) Production of ascites....... 69 (2) Ammonium sulphate precipitation................... 70 (3) Ion exchange chromatography (DEAE)................ 70 (4) Determination of Purity: SDS-PAGE................. 71 2.11 Paraffin histology 2.11.1 Hematoxylin and eosin staining................. 75 2.11.2 Masson trichrome staining...................... 75 2.12 Monoclonal antibodies (mAbs) for in vivo use 2.12.1 Anti-gp39 mAb (MR1)............................ 76 2.12.2 CTLA4-Ig....................................... 76 2.13 Animal related techniques 2.13.1 Anaesthesia.................................... 77 2.13.2 Blood collection............................... 77 2.13.3 Intravenous injection.......................... 78 2.13.4 Intraperitoneal injection...................... 78 2.13.5 Recipient pretreatment......................... 78 2.13.6 Skin grafting.................................. 80 2.13.7 Heterotopic vascularized cardiac grafting (1) Introduction...................................... 80 (2) Recipient preparation............................. 81 (3) Donor operation................................... 82 (4) Implantation of the vascularised heterotopic heart graft....................................... 83 (5) Recovery.......................................... 84 2.13.8 Second cardiac allografts in the neck.......... 85 2.13.9 Assessment of graft survival................... 85 2.13.10 Adult thymectomy.............................. 85 2.13.11 Statistical analysis.......................... 86 2.14 Figures.......................................... 87 Chapter 3: Characterization of resting B (rB) cells in vitro 3.1 Introduction 3.1.1 Aim of this chapter.............................. 89 3.1.2 Background....................................... 89 3.2 Results 3.2.1 FACS analysis of rB cells 3.2.1.1 Expression of B7-1 and B7-2.................... 91 3.2.1.2 Expression of B7-1 and B7-2 after LPS activation .................................... 92 3.2.1.3 Expression of B7-1 and B7-2 after stimulation of allogeneic T cells............................. 92 3.2.2 MLC assay 3.2.2.1 After each step in the preparation for B cell.. 93 3.2.2.2 MLC response of the 60/70% interface without sequential Sephadex G-10 columns....... 94 3.2.2.3 The effect of radiation on rB cells............ 94 3.3.3.4 Comparison of the capacity of rB cells, aB cells and LODACs to act as APCs in MLC...... 94 (a) A single miH barrier, H-Y.......................... 95 (b) Multiple miH barrier............................... 95 (c) A single MHC barrier............................... 95 (d) Multiple miH+MHC barrier........................... 95 3.2.2.5 The effect of preculture with allogeneic rB cells.......................................... 96 3.2.3 CTL assay........................................ 97 3.2.4 ELISA............................................ 97 3.3 Discussion......................................... 98 3.4 Summary........................................... 102 3.5 Figures and Table................................. 103 Chapter 4: Intravenous injection of allogeneic resting B cells 4.1 Introduction 4.1.1 Aim of this chapter............................. 117 4.1.2 Background...................................... 117 4.2 Results 4.2.1. Skin graft prolongation following pretreatment with donor rB cells 4.2.1.1 Immunogenicity of the H-Y antigen............. 119 4.2.1.2 Induction of unresponsiveness to H-Y.......... 119 4.2.1.3 Second skin grafts in H-Y mismatched recipients.................................... 120 4.2.1.4 H-Y mismatched skin grafts in sensitized recipients...................... 121 4.2.1.5 Induction of unresponsiveness to multiple miH antigens and MHC antigens..... 121 4.2.2 Heart graft experiments following pretreatment with donor rB cells 4.2.2.1 Timing of pretreatment with DST............... 123 4.2.2.2 Dose of rB cells required to induce unresponsiveness ................... 123 4.2.2.3 Unresponsiveness to H-Y mismatched cardiac allografts ......... 124 4.2.2.4 Tolerance to H-Y.............................. 125 4.2.2.5 Induction of unresponsiveness to multiple miH antigens ................................. 125 4.2.2.6 Induction of unresponsiveness to a single MHC class I antigens.......................... 126 4.2.2.7 Induction of unresponsiveness to multiple miH+MHC antigens ............................. 126 4.2.3 Characterization of resting B cells after injection 4.2.3.1 FACS (1) Detection of donor cells after injection.......... 128 (2) Expression of B7-2 on donor cells after injection into fully allogeneic recipients (a) Donor cells gated on MHC class II molecules....... 129 (b) Donor cells stained with CFSE before injection ... 129 (3) Expression of B7-2 on B cells after injection in H-Y mismatched recipient Donor cells stained with CFSE......................................... 130 4.2.3.2 Responsiveness of recipient T cells after pretreatment with rB cells (1) MLC............................................... 131 (2) CTLp.............................................. 131 4.3 Discussion........................................ 132 4.4 Summary........................................... 137 4.5 Figures........................................... 138 Chapter 5: Blockade of gp39 / CD40 pathway 5.1 Introduction 5.1.1 Aim of this chapter............................. 159 5.1.2 Background...................................... 159 5.2 Results 5.2.1 Pretreatment with donor cells in combination with MR1 and/or CTLA4-Ig 5.2.1.1 Pretreatment with rB cells and MR1............ 161 5.2.1.2 B7-2 expression on rB cells after MR1 treatment................................. 161 5.2.1.3 Pretreatment with rB cells and MR1 + CTLA4-Ig................................ 162 5.2.1.4 Pretreatment with MHC class II positive cells and MR1........................ 162 5.2.1.5 Pretreatment with MHC class II negative cells and MR1........................ 163 5.2.1.6 Operational tolerance......................... 164 5.2.2 CD40 knockout mice as B cell donors............. 165 5.2.3 gp39 knockout mice as recipients................ 166 5.2.4 CD40 expression on activated T cells............ 167 5.2.5 In vitro analysis of mice pretreated with alloantigen in combination with MR1 5.2.5.1 Induction phase (1) MLC assay......................................... 168 (2) CTLp assay........................................ 169 5.2.5.2 Maintenance phase (1) MLC assay......................................... 169 (2) CTL assay......................................... 170 (3) Alloantibody assay................................ 170 5.3 Discussion........................................ 171 5.4 Summary........................................... 176 5.5 Figures........................................... 178 Chapter 6: Conclusions Discussion, implications of results and a potential strategy to induce donor specific unresponsiveness in clinical transplantation 6.1 Introduction...................................... 195 6.2 Brief summary of results 6.2.1 Brief summary for Chapter 3..................... 196 6.2.2 Brief summary for Chapter 4..................... 196 6.2.3 Brief summary for Chapter 5..................... 197 6.3 Potential strategy for the induction of tolerance in clinical transplantation........................... 198 References............................................ 200 |
Chapter 1: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Introduction 1.1 Clinical aspects 1.1.1 Introduction Despite improvements in medical therapy, organ transplantation continues to be the only effective treatment for patients with end-stage organ failure. Although mechanical dialysis systems have been used to help end-stage renal failure patients, other artificial organs are still far from clinical use with the possible exception of a prototype of an artificial heart = (1)= . Recent pharmacological advances in non-specific immunosuppressive drugs such as cyclosporine = (2)= , methylprednisolone = (3)= , azathioprine = (4)= and polyclonal antilymphocyte globulin = (5)= , as well as technical advances in organ transplantation have improved the outcome of kidney, heart, liver, lung, and pancreas allografts over the last decade = (6-8)= . Although systemic immunosuppression for organ transplantation has improved considerably, its long-term use often results in drug toxicity and cardiovascular disease, and excessive use of non-specific immunosuppressive drugs is a major cause of morbidity and mortality among transplant patients due to the emergence of infectious disease = (9)= and cancer = (10, 11)= . It has long been recognized that these complications could be avoided if it were possible to achieve a condition of donor-specific unresponsiveness, or immunological tolerance to the donor alloantigens. 1.1.2 Historical development Transplantation of organs for the treatment of either acute or chronic organ failure has been an attractive concept for nearly a century. The first experimental kidney transplants in animal were reported by Emerich Ulmann from Vienna in 1902 and Alexis Carrel from America in 1905. The first attempts at human kidney transplants were in 1902 by Ulmann and 1906 by Jaboulay. These early experiments simply established that such kidney transplants were technically possible. However, the problem of rejection was first highlighted in 1910 when Carrel noticed the better graft survival rate of autografts compared with allografts (then called homografts). Then in 1923, Carl Williamson from the Mayo Clinic clearly defined the histological features of organ rejection in kidney transplantation. In 1946, a human allogeneic kidney anastomosed to arm vessels under local anaesthesia was attempted by three young surgeons, Charles Hufnagel, Ernest Landsteiner and David Hume at the Peter Bent Brigham Hospital in Boston. The transplanted kidney took over until the patient's native kidneys began to function and she made a full recovery. This success led Hume to start a series of cadaver kidney transplants without immunosuppression in 1951. The results were poor with all grafts failing except for one patient whose transplant functioned for six months. It was then realised that if organ transplantation were to be successful, the problem of rejection needed to be solved. In 1953, Morton Simonsen in Copenhagen and William Dempster in London defined the problems of kidney rejection in dogs. Around the same time, Rupert Billingham, Leslie Brent and Peter Medawar showed that foetal and neonatal mice injected with donor cells were able to accept skin grafts from the donor = (12)= , demonstrating for the first time that it was possible to transplant tissue between unrelated individuals. For allografts, suppression of the immune response was necessary. Murray in Boston, K殱s from Paris, Jean Hamburger, also from Paris and Shackmann in London employed total body irradiation but the mortality rate proved to be too high = (14)= . The first major breakthrough in immunosuppressive agents came in 1959, with the discovery of 6-mercaptopurine by Schwartz and Damesheck, which was subsequently shown by Roy Calne, Charles Zukoski and David Hume to prolong kidney allograft survival in dogs = (15)= . Azathioprine, which is metabolised to 6-mercaptopurine, was introduced in 1962 = (16)= and is still being used routinely today, in combination with steroids and cyclosporine A = (17-19)= . Cyclosporine A was discovered by Jean Borel in Basle = (20)= and introduced into clinical use, again by Calne together with his colleague David White = (2)= . Since the success of kidney transplantation, many other organs can now be transplanted. The first clinical transplant of a bone marrow was performed by G. Mathe in 1958 = (21)= ; of a liver by Thomas Starzl in 1963 = (22)= ; and of a heart by Christian Barnard in 1967 = (23)= . 1.1.3 Current immunosuppression Since the first successful human renal transplant in the 1946 (see previous), graft survival rates have improved to between 80 and 90% after the first year = (8)= . However, chronic immunosuppression of the recipient is required for long term survival of the graft. Today, azathioprine, cyclosporine A and steroids are routinely used in the clinic = (17-19)= . New agents are becoming available, for example, Tacrolimus (FK506) = (24, 25)= , Mycophenolate mofetil = (26)= , Rapamycin = (27)= , 15-Deoxyspergualin = (28)= , Leflunomide = (29)= and Brequinar sodium = (30)= . For acute rejection, more aggressive treatments such as anti-lymphocyte globulin, anti-thymocyte globulin, OKT3 and high dose steroids are available. Monoclonal antibodies (mAb) are also being introduced into clinical use. At present OKT3 (anti-CD3) is the only mAb licensed for human use = (31)= but other mAb are being tested. These include anti-CD4 mAb and anti-CD25 (IL2 receptor) mAb. All these agents have non-specific immunosuppressive effects, increasing the susceptibility of transplant recipients to opportunistic infections and neoplasia = (32, 33)= as well as giving rise to many other unwanted side effects such as marrow suppression, hypertension, tremor, gingival hypertrophy, hyperlipidaemia, diabetes and osteoporosis. Furthermore, current immunosuppression does not prevent chronic rejection of transplanted organs, leading to 5% graft loss per year in renal transplantation, a problem that is largely unresolved at present = (34)= . 1.2 Graft rejection 1.2.1 Immune recognition The immune system in mammals has evolved to recognize and destroy the large variety of potential pathogens such as viruses, bacteria and parasites, which an individual may encounter. Recognition is mediated by T and B lymphocytes. B cells have surface immunoglobulin (Ig) as their specific antigen receptor and T cells the T cell antigen receptor (TCR). The two types of receptors are structurally distinct and are generated by different sets of genes. T cells usually recognize processed or degraded antigen, and only when it is physically associated with molecules encoded by the major histocompatibility complex (MHC) = (35-38)= . In effect, MHC molecules act as a guidance system for T cells = (39)= , allowing them to recognize antigens from within cells. These three groups of molecules, the antibodies, the TCRs and the MHC molecules, control the process of immune recognition. 1.2.2 Immune rejection of allografts The immunological nature of tissue rejection was firmly established over 50 years ago when Medawar et al. found that skin grafts between genetically disparate humans or rabbits underwent rapid necrotic changes and were sloughed off. This rejection process displayed marked specificity and memory for donor tissue and was accompanied by infiltration with leukocytes = (40, 41)= . It has been clear for many years that T lymphocytes play an essential role in graft rejection because both congenitally athymic nude mice = (42-44)= and T-cell-deprived animals fail to reject either allografts or concordant xenografts. Although T cells are required for graft rejection, the relative contribution of the helper and cytotoxic populations is controversial. Two basic mechanisms have been proposed although they are not mutually exclusive. One mechanism is a direct destruction of the grafted tissue by specific cytotoxic T cells. In vitro, MHC mismatched lymphocytes proliferate in response to alloantigen resulting in the generation of T lymphocytes that will lyse target cells of the MHC mismatched type = (45, 46)= . This lysis, or cytotoxicity, is measured in vitro by assessing the release of radioactivity from 51Cr-labelled target cells = (45)= . However, clear evidence implicating such cytotoxicity in vivo has been lacking. Probably the clearest demonstration is that cloned populations of donor-specific cytotoxic T lymphocytes (CTL) have been used to mediate both tumor graft = (47)= or skin destruction = (48)= . Evidence that the rejection response was specific was obtained in experiments using allophenic mice = (49)= . B6ォA/J allophenic mice are genetic mosaics whose individual cells express H2b or H2a histocompatibility antigens but not both = (50)= . Trunk skin from B6ォA/J allophenic mice was grafted onto immunoincompetent H2b nude mice and allowed to heal and regrow hair that was both black and white, reflecting the genetic mosaicism of the allophenic grafts. One month after engraftment, the H2b nude animals were reconstituted with syngeneic H2b T cell reactive against H2a allodeterminants. Although the intensity of the nonspecific inflammatory response induced destruction of the epidermis and complete hair loss, the allophenic grafts survived and regrew hair and the predominant colour of that hair was black. This experiment showed that the final stage of the rejection could be specific for its target, suggesting that cytotoxic T cells might be involved in the final stage of graft rejection. The second mechanism is a "delayed-type hypersensitivity (DTH)-like" process, in which CD4+ T cells may be sensitized against MHC class II molecules presented by the few dendritic leukocytes from the graft. The sensitized T cells then secrete lymphokines that recruit and activate macrophages which damage the graft tissue. In this mechanism, the final stage of destruction is non-specific since lymphokines and macrophages can not recognize donor antigens. Support for the role of DTH reactions in graft rejection comes from a study investigating the rejection of H-Y disparate grafts. While B10.A(5R) (KbIAbIEb/kDd) female mice rejected syngeneic male skin grafts, they failed to generate cytotoxic T cell responses to H-Y in vitro = (51)= , suggesting that cytotoxic T cells were not essential to induce graft rejection. Although clear evidence that DTH responses are responsible for graft rejection has been lacking, skin graft studies have shown that CD4+ T cells play a central role in graft rejection in the mouse = (52)= and the rat = (53)= . These studies have shown that adoptive transfer of CD4+ T cells, but not CD8+ T cells, restores skin graft rejection in T-cell-deprived animals. This type of adoptive transfer study has been extended by other workers to examine the cellular requirements for experimental heart and kidney allograft rejection = (54-56)= . The finding that CD4+ T cells are central to the initiation of graft rejection are somewhat confused by demonstrations that, in certain mutant strains which differ only by a few amino acids in their MHC class I molecules, only the CD8+ T cells are required to restore graft rejection = (57)= . This suggests that in some situations class I-restricted CD8+ helper T cells are generated = (58, 59)= . It is now widely accepted that the immune response that mediates allograft rejection is initiated by the recognition of foreign MHC molecules = (59)= . However, the alloantibody response and natural killer (NK) cell activities may also be involved in graft rejection. Preformed antibodies directed at MHC antigens resulting from previous exposure to MHC-incompatible tissue (positive crossmatch) induce hyperacute rejection in man and natural antibodies to xenoantigens induce hyperacute rejection of xenografts = (60-62)= . The role of alloantibodies in acute or chronic allograft rejection is not well understood = (63)= . 1.2.3 Direct and indirect pathways of allorecognition The response of T cells to alloantigen has been demonstrated to result from the recognition of intact allo-MHC molecules (+ peptide) present on either the allograft itself or on donor passenger leukocytes (Figure 1-1). This form of recognition has been termed the direct recognition pathway and is thought to represent the primary way in which the immune system "sees" alloantigen in acute allograft rejection. However, a second pathway of recognition of alloantigen has also been identified and is termed the indirect pathway (Figure 1-1) = (71, 72)= . This indirect pathway involves the presentation of peptides derived from donor-MHC molecules by recipient antigen presenting cells (APCs). The involvement of this pathway in allograft rejection has been inferred from data demonstrating that recipient T cell-depleted leukocytes, previously sensitized to donor alloantigens, can transfer sensitization to second naive recipients. This effect was caused by the active sensitization of the second hosts and not by contaminating donor strain cells = (73)= . Clear evidence of a role for indirect presentation in vivo was obtained by Benichou et al.. They collected T cells from mice which had been sensitized by infusion of allogeneic splenocytes or a skin graft. They then demonstrated proliferation to synthetic peptides derived from polymorphic regions of both the a and b chains of the donor IAk molecule presented by host APCs = (74)= . In another study, when skin from MHC class II-deficient mice was transplanted to normal allogeneic recipients, the rejection time was not different to MHC expressing grafts, demonstrating that MHC class II antigens need not be expressed on a donor graft for rapid rejection to occur = (75)= . This observation is significant because in the same study it was also shown depletion of CD4+ cells resulted in graft prolongation. These results suggested that donor (MHC class I) antigens present in the graft were recognized in association with recipient MHC class II molecules by CD4+ T cells, and that this indirect recognition pathway was sufficient for initiating the rejection of the skin graft = (76)= . Evidence that the indirect pathway might be important in chronic or delayed graft rejection was obtained by Braun et al. = (77)= who showed that the direct pathway was only operated when donor passenger leukocytes were still present in the graft. A T cell clone which recognized donor antigen directly was found to reconstitute rejection of an allograft in an empty rat. However, if graft was depleted of passenger APCs, the T cell clone could not induce rejection. 1.2.4. H-Y antigen In the 1930s and 40s Peter Gorer and George Snell found that MHC antigen(s) were involved in the rejection of transplanted tumours and other tissues = (78)= . Subsequently typing for antigens has improved the success rate of transplantation = (79)= . However, even if MHC antigens are identical between donor and recipient, rejection still occurs as minor histocompatibility (miH) antigens are also immunogenic = (80, 81)= . One of the best characterized miH antigens, H-Y, was originally discovered as a transplantation antigen by Eichward and Silmer in the mid-1950s = (82)= . Females of the H2b strain, C57BL/6, rapidly rejected primary syngeneic male skin grafts = (83, 84)= . However, not all recipients respond to an H-Y mismatch. H2 congenic strains such as B10.D2 (H2d) and B10.BR (H2k), differing by an allelic segment of H2, were found to be non-responders to H-Y as were non-H2b strains tested on several genetic backgrounds. These results implicated H2 gene(s) in the context of immune responses = (83)= . We now know that miH antigens such as H-Y are recognized by T cells as peptides bound to an MHC molecule. The ability of an APC to process and present H-Y will depend on both the MHC class II genes encoding the IA and IE molecules together with genes involved in antigen processing including H-2M and H-2O .= (85-87)= 1.3 Mechanisms of tolerance 1.3.1 Historical perspective of immunological tolerance Owen introduced the concept of immunologic tolerance 50 years ago with his studies on chimerism in dizygotic twin cattle = (88)= . Owen described tolerance as the absence of an immune response against a normally foreign cell to explain his failure to detect cytotoxic antibody responses against the co-twin's red blood cells. Billingham, Brent, and Medawar reported subsequently that immunologic tolerance against foreign skin grafts could be achieved in mice if the mice were previously exposed to the foreign cells during the foetal or (less often) the neonatal period = (12)= . Shortly afterward, Burnet proposed the clonal selection theory based mainly on studies of B cells and antibody responses, and theorized that the immune system eliminated clones of B cells with receptors specific for self-molecules during lymphoid development = (89)= . As a result of early studies of neonatal tolerance, three important ideas emerged and formed the theoretical basis of immunologic tolerance. (i) The immune system developed to distinguish primarily between self protein and non-self proteins. (ii) The immune system restricted the education of self versus non-self to the foetal/neonatal period. (iii) Tolerance (specifically neonatal tolerance) involved mainly deletion mechanisms in thymus. Although the details and specifics of these three statements have been challenged in the intervening years, the basic ideas underlying these hypotheses have been accepted as dogma. However, Matzinger has recently proposed the new viewpoint that the immune system does not discriminate between self and non-self, but that its primary driving force is the need to detect and protect against danger = (90)= . 1.3.2 Deletion of self-reactive T cells In the thymus, rapidly proliferating T cell precursors rearrange their TCR genes = (91)= . T cells are positively selected based on the specificity of their randomly created antigen receptors. T cells restricted to self MHC molecules survive (positive selection) and become selected into CD4+ or CD8+ subsets depending on whether they are restricted to MHC class II or class I molecules, respectively. It is at this stage of development that self-reactive T cells are deleted (negative selection) = (92-96)= . Before the mid-1980s, it was difficult to distinguish experimentally between deletion, anergy or active suppression as the mechanisms of self-tolerance because potentially self-reactive T cells could not be identified directly. More recently, the development of TCR-specific mAbs and TCR transgenic mice have helped in the understanding of these processes. A set of mAbs specific for individual TCR Vb regions were used to detect clonal deletion in vivo. Kappler et al. showed that, in IE-expressing mice, thymocytes bearing the Vb17a which confer reactivity for IE molecules were present at almost normal levels in the CD4+CD8+CD3low population, but were deleted from mature thymocytes and from peripheral T cell populations = (93)= . In another series of experiments, rearranged TCR a and b genes were isolated from a T cell clone which recognized the male-specific transplantation antigen H-Y in the context of H2b. These genes were injected into fertilized mouse ova to create transgenic mice. In H2b TCR transgenic females, the thymus contained the normal CD4 and CD8 defined cell populations. However, in H2b transgenic males which expressed not only the transgenic TCR but also the target H-Y antigen, the thymus was small and most of the thymocytes were CD4-CD8- = (92)= . Both experimental approaches revealed that self-reactive T cells were deleted in the presence of their specific self-antigen and in both situations, deletion of self-reactive T cells occurred in the thymus. 1.3.3 The existence of self-reactive T cell in the periphery Although clonal deletion in the thymus has clearly been demonstrated in a number of different models = (97)= it is not the only mechanism that prevents self-reactivity. Several experiments have shown that self-reactive T cells can co-exist with their cognate self-antigens in the periphery without resulting in autoimmunity = (98, 99)= . Such observations together with the fact that many tissue specific self-proteins may not be expressed in the thymus have suggested that other mechanisms of tolerance must exist in the periphery. Alternatively, T cells with low affinity for self-antigen may escape deletion = (100, 101)= , but are unable to achieve a high enough affinity/avidity to become activated. This state of functional inactivation is termed anergy and was clearly demonstrated in minor lymphocyte stimulatory (Mls)-1b mice injected with foreign cells expressing the Mls-1a gene. In this experiment, Vb6 T cells which should react to Mls-1a were not deleted, but responsiveness to Mls-1a was abolished = (102)= . 1.3.4 History of Anergy, "functional deletion" In 1970, Bretscher and Cohn proposed the concept of functional deletion, or "paralysis," when they introduced the model that B cells required two signals in order to become activated. They reasoned that the immune system had to develop mechanisms of silencing potentially autoreactive B cells responses that arise during adulthood, resulting from somatic mutation in B cells. They proposed that the immune system recognized two separate antigenic determinants on any antigens and that a B cell required two separate signals to become fully activated. Recognition of antigen by the B cell receptor (surface Ig) alone sent a signal that paralyzed the cell (signal 1). They proposed that the delivery of signal 1 plus the simultaneous recognition of a second signal was required to induce a fully activated B cell response = (103)= . These observations and their interpretation by Bretscher and Cohn provided the groundwork for Lafferty to expand the two-signal hypothesis to the T cell compartment. Lafferty proposed from his studies of tissue transplantation that T cells required not only the recognition of antigen (signal 1), but also some other signals from an APC (signal 2) in order to become fully activated (Figure 1-2) = (104)= . Because hematopoietic cells (but not other cell types) could stimulate allogeneic responses in vitro, he proposed that hematopoietic cells possessed the capacity to provide a necessary co-stimulatory signal to achieve full T cell activation. Lafferty demonstrated that culture of thyroid grafts for several days prior to transplantation resulted in prolonged survival and noted that in vitro-cultured organs contained very few leukocytes = (105)= . Lafferty postulated that the depletion of "passenger" leukocytes from the donor tissue eliminated the co-stimulatory ability of the graft. About 10 years after this hypothesis was proposed, clear evidence for T cell inactivation following "defective" antigen presentation has come from several experiments. Lamb and Feldmann found that high concentrations of antigen in the absence of APCs could inactivate human helper T cell clones = (106)= . Schwartz et al. showed that APCs fixed by ECDI (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide) = (107)= or purified MHC class II molecules incorporated into planar membranes = (108)= pulsed with peptide antigen induced a similar anergic state in mouse helper T cell clones. After exposure to ECDI fixed APC or MHC class II on planar membranes, T cell clones were found to be unresponsive to a subsequent stimulus by fully competent APC. Also, in vivo, fixed APCs have been demonstrated to induce tolerance in normal T cells = (107)= . Gaspari et al. found that keratinocytes expressing MHC class II molecules induced unresponsiveness in T cell lines = (109)= , while Markmann et al. showed that pancreatic b cells expressing transgenic MHC class II molecules also induced unresponsiveness = (110)= . Cho et al. have shown that anergic T cells are unable to make enough IL-2 to support their own proliferation. If IL-2 was added, T cell responses were restored = (111)= . Among a number of possible candidates CD28 was considered to be a very important provider of second signals but it was not until the experiments of Harding and Allison, using the same ECDI system as Jenkins and Schwartz, that it was demonstrated that anti-CD28 mAbs could substitute for APC delivered costimulation = (112)= . Such experiments have led to the concept that the CD28/B7 pathway is probably the most important provider of second signals. Other candidates for the so-called accessory or second signals involved in the APC-T cell interaction are thought to be cell surface molecules including CD4 and CD8 that participate in recognition of MHC class II and class I molecules respectively, as well as the leukocyte function antigen (LFA)-1/ intracellular cell adhesion molecule (ICAM)-1 = (113)= , CD2/LFA-3, and gp39/CD40 interactions = (114)= . 1.3.5 Deletion and anergy of B cells Nemazee and Burki provided evidence of B cell clonal deletion. They generated mice carrying transgenes for the heavy and light chain of an antibody reactive with H2k MHC class I antigens (anti-H2-KkDk). While 20 to 50% of B cells expressed the Ig transgenes in H2d transgenic mice, these cells could not be detected in the (H2d x H2k) F1 transgenic mice, supporting a role for deletion in B cell tolerance = (115)= . On the other hand Goodnow et al. have shown evidence of B cell clonal anergy. They constructed mice carrying a transgene for the antigen hen egg lysozyme (HEL) and other mice carrying transgenes for a high-affinity anti-HEL antibody in which 90% of B cells expressed sIgM and sIgG with specificity for HEL. The two strains of mice were crossed to provide progeny which expressed both HEL and anti-HEL sIg. B cells from these mice were not deleted but could no longer secrete anti-HEL antibody when challenged with HEL in the presence or absence of T cell help = (116)= . They also showed that transferring anti-HEL transgenic splenic B cells into HEL transgenic mice rapidly induced clonal anergy and down-regulation of sIgM = (117)= and that clonal anergy in B lymphocytes could be broken under particular conditions such as bacterial lipopolysaccharide (LPS) stimulation = (118)= . 1.3.6 Evidence for an active mechanism of tolerance Deletion and anergy are thought to be passive mechanisms of tolerance. Several experimental findings have recently suggested an active regulatory mechanism in models of peripheral tolerance to alloantigens. Firstly, tolerance depends on the persistence of antigen = (119, 120)= . Hamano et al. have shown that mice treated with anti-CD4 mAb at the time of transplantation accepted fully allogeneic cardiac grafts indefinitely, and second donor type hearts were accepted at any time after transplantation. However, when original grafts were removed 50 days after transplantation, recipients were able to accept second donor type hearts 60 or 130 days later but by 200 days the second heart grafts were rejected = (120)= . Therefore, the absence of grafts in tolerant recipients for 150 days restored the ability to mount a rejection response. Secondly, once tolerance is induced, even the injection of large numbers of naive non-tolerant T cells can not break the tolerant state = (121, 122)= . Thirdly, tolerance can be adoptively transferred to naive animals, usually by CD4+ T cells = (122-127)= . Fourthly, cellular infiltrates are found frequently in nonrejected grafts as well as in tissues protected from autoimmune destruction = (66, 128)= . Finally, it has been shown that cells from tolerant recipients can prevent proliferation or the generation of cytotoxic responses specific for donor alloantigens = (129)= . Moreover, recently T cells have been shown to have regulatory activity. Fowell and Mason have demonstrated that radiation induced diabetes in the rat can be prevented by adoptive transfer of CD45RClow cells = (130)= . In the mouse, wasting disease induced by CD45RBhigh CD4+ T cells can be prevented by CD45RBlow CD4+ T cells = (131)= and colitis induced in C.B-17 scid mice by injection of CD45RBhigh cells can be prevented by co-transfer of CD45RBlow cells = (132)= . Groux et al. have shown that CD4 T cell clones polarized in vitro with IL-10 inhibited antigen-specific T-cell responses in vivo = (133)= . Taken together, these observations imply that regulatory cells might be involved in protecting against tissue destruction and that peripheral tolerance might involve the generation of active regulatory mechanisms. 1.3.7 Infectious tolerance The clearest example of a peripheral active mechanism of tolerance in transplantation is so called "infectious" tolerance, which was demonstrated very elegantly by Qin et al. = (122)= . CBA mice transgenic for human CD2 were thymectomized and made tolerant of allogeneic B10.BR skin using a short-term treatment of non-depleting CD4 and CD8 mAbs. The tolerant T cells in these mice could be identified by the expression of the human CD2 transgene. When 50 million normal (hCD2 negative) spleen cells were infused into the tolerant recipients, the skin grafts were maintained despite clear donor T-cell chimerism. It could be shown that the naive T cells had the potential to reject the graft by depleting the hCD2 tolerant cells at the time of naive cell infusion. However, after 2 weeks of co-existence with tolerant T cells and the graft, the infused cells became tolerant in their own right. This could be shown by depleting the original tolerant population using anti-human CD2 mAb. The mice remained tolerant of allogeneic skin if challenged with fresh grafts, even though no original human CD2+ cells could be seen by flow cytometry. 1.3.8 Immune deviation (Th1/Th2 paradigm) There are now many examples where the immune response can be considered as a polarised differentiation of the CD4+ T-cell compartment towards that exemplified by T-cell clones expressing different patterns of cytokines in vitro = (134)= . Cell-mediated responses including DTH, graft rejection, certain autoimmune diseases, and complement-fixing antibody responses to bacteria, are thought to be mediated by T cells making inflammatory cytokines such as interferon-g (IFN-g) and interleukin (IL)-2 similar to Th1 clones. Conversely, humoral responses, especially those involving neutralising IgG1 or antiparasitic IgE antibodies, are thought to be mediated by Th2 clones secreting IL-4, 5, 6 and IL-10. Initially, following antigen recognition naive CD4+ T cells are thought to correspond to Th0 clones in that they simultaneously make low levels of both Th1 and Th2 type cytokines, but as the response develops the Th0 cells, begin to polarise towards either a Th1 or Th2 phenotype. This is reinforced as each subset has an autocrine response to its own pattern of cytokines that act further to suppress the development of the opposite subset = (135)= . In some cases tolerance can be achieved by deviation of an aggressive Th1 response into a relatively innocuous Th2 one = (136-138)= . Dominant Th2-like responses have been observed in oral tolerance to protein antigens = (139, 140)= or in neonatal tolerance to transplantation antigens = (138, 141-144)= . A number of groups have also shown preferential expression of IL-4 and/or IL-10 during the induction of tolerance in rats or mice to renal or cardiac grafts = (125, 145)= . In some rodent models it is even possible to modulate or block the induction of autoimmune disease = (146)= , graft versus host disease = (147)= , or skin graft rejection by the transfer of T cells expressing Th2 type cytokines = (148)= . It is therefore clear that apparent deviation of an immune response is one possible route to the induction of certain forms of tolerance. Despite the attractiveness of the Th1/Th2 paradigm to explain tolerance there are very few data to support the idea that Th2 response induce tolerance directly = (149)= . 1.3.9 T cell ignorance One other possible mechanism for the induction of specific unresponsiveness could be that T cells are simply unaware or ignorant of the presence of alloantigen. The concept of T-cell ignorance was originally proposed by Ohashi et al.. The authors generated mice in which the expression of lymphocytic choriomeningitis virus (LCMV) glycoprotein was directed to the ァ cells of the pancreas using the insulin promoter as a transgene. In these mice the LCMV glycoprotein was simply ignored by the immune system evoking neither activation nor tolerance in T cells. However, immunization of mice in which a very large proportion of T cells expressed the LCMV glycoprotein-specific TCR, with LCMV led to a specific distruction of the pancreas = (150)= . Similar observations have been made in mice transgenic for the MHC class I alloantigen, H2-Kb. The works of Morahan et al. = (99)= and Allison et al. = (151)= , using a model in which tolerance to H2-Kb was elicited by introduction of H2-Kb as a transgene under control of the insulin promoter, was initially taken to support the clonal anergy theory of peripheral tolerance. These data have been reinterpreted in the light of evidence that this transgene is in fact expressed at low levels in the thymus, as well as in b cell of pancreas. These workers have now suggested that high-affinity H2-Kb-specific cells are deleted in the thymus, leaving a population of cells which is able to cause rejection of H2-Kb bearing skin, but ignores the same antigen when expressed on b cells. However, the presence of high levels of IL-2 in the b cells was able to reverse this nonchalant attitude amongst H2-Kb-specific cells which then destroyed the ァ cells = (152)= . 1.4 Antigen presenting cells (APCs) 1.4.1 Historical context As T cell activation is a key event in initiating graft rejection, an understanding of the cellular mechanism integral to the recognition of alloantigen by T cells has been one of the major goals of transplant immunology. Early studies demonstrated that exposure of purified primed T cells to antigen alone in vitro failed to induce either proliferation or "help" for B cell antibody production = (153, 154)= . These responses were restored by adding back an autologous fraction of glass-adherent, non-lymphoid cells. They were called "accessory cells". Dissociated spleen cells also made antibodies when they were incubated with antigen and washed to remove the excess, despite only a small amount of antigen remaining cell-associated, suggesting that antigens might be taken up within some cells. Antigen-pulsed accessory cells were also effective in stimulating lymphocyte responses, but pulsed fractionated-lymphocytes were not. Therefore, antigen seemed to be "processed" to a more stimulatory form by accessory cells, and it was suggested that antigens were endocytosed by accessory cells before being processed and presented to specific lymphocytes. Thus, accessory cells also came to be known as APCs. The discovery of MHC restriction led to the idea that processed antigens become associated with MHC class II molecules on accessory cells and stimulated helper T cells. Since MHC class II-restricted helper cells are often required for the development of effector cells, APCs were generally thought of as primarily MHC class II-bearing cells. From these studies came the concept that there existed specialized APCs whose function was to deliver foreign antigen to helper/inducer T cells in immunogenic form. To qualify as a "professional APC", a cell must fulfill at least three distinct functions: (i) uptake and processing of antigen, (ii) expression of the antigen on the cell surface in the context of MHC-encoded class II molecules, and (iii) provision of costimulatory signals. Based on in vitro model systems, among the cells that are likely to be important for T cell activation under normal conditions are MHC class II-bearing monocytes/macrophages and dendritic cells = (155)= . 1.4.2 Dendritic cells Adherent accessory cells from lymphoid tissues (e.g. mouse spleen) were found to contain a distinct cell type in addition to the more numerous macrophages = (156)= . These cells were irregular in shape and actively produced and retracted cell processes, in contrast to macrophages which were more sessile and rounded. They were termed lymphoid dendritic cells. Mouse splenic dendritic cells only transiently adhere to tissue culture plastic, have a low buoyant density, and lack Fc receptors, so they can be separated from macrophages which are more adherent, have a higher density and form rosettes with antibody-opsonized particles. Dendritic cells are the most effective APCs at activating naive CD4+ T cells and are approximately 10-100 fold more efficient at inducing T cell proliferation than unfractionated allogeneic splenocytes = (157)= . Moreover, dendritic cells are able to stimulate proliferation of both resting and activated T cells while other APCs such as macrophages and aB cells are thought to be capable of stimulating only T cells which have already been activated = (155)= . Dendritic cells are found in many nonlymphoid tissues but can migrate via the afferent lymph or the blood stream to the T-dependent areas of lymphoid organs = (158, 159)= . After foreign proteins are administered in situ, dendritic cells are a principal reservoir of immunogen. In vitro studies indicate that dendritic cells only process proteins for a short period of time, when the rate of synthesis of MHC products and content of acidic endocytic vesicles are high = (160)= . Antigen processing is selectively dampened after a day in culture, but the capacity to stimulate responses to surface bound peptides and mitogens remains strong = (161, 162)= . Dendritic cells are motile, cluster efficiently = (163, 164)= and activate T cells that are specific for stimuli on the cell surface = (165)= . 1.4.3 B cells as APCs As MHC class II molecules are also expressed on B cells, the ability of B cells as APC has been investigated. One of the first experiments demonstrating antigen presentation by B cells was reported by Chesnut and Grey who developed a model system to determine whether B cells could present antigen bound by cell surface Ig. Whereas T cells from mice primed with rabbit Ig failed to respond to B cell-enriched (adherent cell depleted) APCs plus normal rabbit IgG, these T cells proliferated vigorously to rabbit anti-mouse Ig in the presence of B cell-enriched APCs, suggesting that B cells used surface Ig to take up antigens. Both normal rabbit IgG and rabbit anti-mouse Ig were equally stimulatory when plastic-adherent cells (macrophages and dendritic cells) were used as APCs = (166)= . In a subsequent study, it was reported that normal resting B (rB) cells were poor at presenting typical soluble antigen, such as Keyhole limpet hemocyanin, to antigen-specific T cells whereas LPS-stimulated B cell blasts were effective = (167)= . These studies were integrated by the suggestion that in the initial report rabbit anti-mouse Ig had been presented by preactivated B cells = (168)= . In terms of the ability of rB cells to present antigen, Ashwell et al. showed that highly purified small rB cells could present a variety of conventional protein antigens to normal T cell clones, T cell hybridomas, and antigen-primed T cells = (169)= . However, the ability of rB cells to function as APCs for naive T cells was very weak. Krieger et al. found that the potency of splenic B cells to act as APCs varied with their size; the smaller the B cell, the less potent = (170)= . Therefore, small rB cells have generally been thought to be incompetent APCs (Figure 1-2). Several experiments have shown that small rB cells induce unresponsiveness or a poor response in naive T cells = (166-172)= . It has been suggested that rB cells may be potent inducers of anergy. For example, Eynon et al. showed that mice injected intravenously with fragments (Fab) of rabbit anti-IgD antibody, which targeted antigen directly to rB cells, induced tolerance to the Fab fragments after secondary challenge with the same antigen = (173-175)= . Therefore, under some conditions rB cells might be tolerogens in vivo. 1.4.4 APC in transplantation APCs are thought to be critical to allograft rejection. Experiments performed in the early 1970s demonstrated that depletion of specialised APC, probably dendritic cells, from allografts by organ culture or irradiation before transplantation led to prolonged allograft survival = (105, 176-180)= . The importance of dendritic cells in graft rejection was also demonstrated by experiments in which donor dendritic cells were exchanged with dendritic cells of recipient type within the graft. This was achieved by placing grafts in immunosuppressed recipients and allowing replacement of APCs to occur by the natural turnover of bone-marrow derived cells. Such grafts transplanted into a secondary recipient showed prolonged survival. Moreover, injection of dendritic cells from the tissue-donor induced a rejection episode in long-term surviving grafts, indicating that long-term engraftment was not due simply to a loss of target structures on the graft but due to a lack of immunogenicity = (72)= . In the early 1980s, several experimental studies showed that removal or inactivation of allostimulatory activity in transferred lymphoid cell populations could enhance their tolerogenic effect. Faustman et al. showed that immunization with donor red blood cells depleted of MHC class II-bearing cells induced unresponsiveness and markedly prolonged the survival of transplants of freshly isolated islets. In contrast, immunization with donor blood containing MHC class II-bearing cells resulted in sensitization of the recipients and acceleration of islet allograft rejection = (181)= . Lau et al. showed that pretreatment, on three occasions (1, 2 and 3 weeks before transplantation), with ultraviolet light irradiated Lewis blood induced indefinite prolongation of pancreatic islets from Lewis but not WF rats, while non-irradiated blood was ineffective. They concluded that inactivation of MHC class II-bearing cells might be just as effective as elimination = (182)= . The identity of the MHC class II+ cells was not determined in either of these studies. In an alternative approach, some investigators have used Sephadex G-10 filtration to deplete accessory cells such as dendritic cells or macrophages but not B cells. Ryan et al. showed that when C3H mice were pretreated with accessory cell-depleted C57BL/10 splenocytes, their peripheral blood lymphocytes of the recipients were rendered specifically unresponsive for the duration of the experiment (13 days), whereas pretreatment with unfractionated donor cells led to hyperresponsiveness = (183)= . This experiment was repeated by Hori et al. across a single MHC class II difference (the bm12 mutation in the MHC class II molecule of B6 mice). A single intravenous (i.v.) injection of accessory cell depleted (B6 x bm12)F1 spleen cells into B6 mice abolished the ability of lymphocytes from the mice to respond to bm12 stimulators in vitro, and moreover, greatly prolonged the survival of bm12 skin grafts = (184)= . In each case, the tolerizing APC for the MHC class II alloreactive T cells was likely to be a B cell since other MHC class II+ cells had been removed. Finally, Fuchs and Matzinger showed directly that purified rB cells could induce unresponsiveness to a single minor alloantigen. As mentioned in Section 1.2.4 C57BL/10 mice respond to H-Y. In this experiment 1x107 rB cells were purified from the spleens of male C57BL/10 mice and injected intravenously into female recipients. Splenocytes from the treated female mice were unable to generate cytotoxic T cells against C57BL/10 male targets but retained responsiveness to B10.A targets. When splenocytes were used for pretreatment instead of rB cells cytotoxic T cells against C57BL/10 male targets were generated. Therefore, i.v. injection of male rB cells into female recipients induced specific tolerance to the H-Y. Next, C57BL/10 female mice were pretreated with either 1x107 rB cells or 3x105 low density adherent cells (LODACs, a population enriched for dendritic cells) and transplanted with a male skin graft one week later. All C57BL/10 female mice pretreated with male rB cells accepted male skin grafts indefinitely, while mice pretreated male LODACs rejected H-Y mismatched grafts within 18 days = (185)= . Tolerance induction by rB cells could explain the beneficial effect of donor-specific blood transfusions, which have accounted for significant improvements in renal transplant success rates in clinical = (186-188)= and experimental studies = (189, 190)= . These findings suggest that in limited situations rB cells may play an important role in the induction of donor-specific unresponsiveness. However, in each of the experimental systems described, only limited alloantigenic differences have been involved. 1.5 Costimulatory molecules 1.5.1 B7-CD28 pathway T cells play a critical role in the initiation and regulation of the immune response to antigen. Both the engagement of the TCR with MHC/antigen and a second costimulatory signal are needed for the complete activation of the T cell (Figure 1-2). The B7/CD28 interaction is one of the dominant costimulatory pathways. Interruption of this signaling pathway with CD28 antagonists not only results in the suppression of the immune response, but in some cases induces antigen-specific tolerance = (191)= . However, the B7/CD28 system is increasingly complex due to the identification of multiple receptors and ligands with positive and negative signaling activities = (192)= . 1.5.1.1 Expression of CD28/CTLA4 cDNAs for CD28 and CTLA4 were cloned by Arufo and Seed = (193)= and Brunet et al. = (194)= respectively. The CD28 glycoprotein is expressed constitutively on the surface of 80% of human T cells (all CD4+ cells and about 50% of CD8+ cells express CD28) and on virtually 100% of mouse T cells = (195, 196)= . CD28 is also highly expressed on developing thymocytes, although its role in T cell development is not well understood = (197, 198)= . CD28 expression is not static because the levels of CD28 increase on T cells following activation = (199)= . However, ligation of CD28 with either anti-CD28 mAb or B7-1-transfected cells in the presence of PMA (phorbol myristate acetate) or PHA (phytohaemagglutinin) stimulation downregulates CD28 mRNA levels and cell surface expression = (200)= . CTLA4, on the other hand, is not constitutively expressed on T cells. Instead, it is upregulated following T cell activation due to positive regulatory elements = (194, 201, 202)= . In both human = (201)= and mouse systems = (203)= , cell surface expression of CTLA4 peaks 48 hours after activation, returning to background levels by 96 hours. Thus, the period of time in which CD28 is transiently downregulated and less responsive to signaling is the time period during which CTLA4 expression is maximal, suggesting that CTLA4 may be functionally active at a time when CD28 function is impaired. The ligation of the CD28 molecule appears particularly important for CTLA4 upregulation and function. Finally, activated CD28 -/- mouse T cells did not express significant amounts of CTLA4 at the cell surface unless exogenous IL-2 was added, suggesting that CD28 regulates CTLA4 expression = (203)= . Integrins may also play a role in the regulation of CTLA4 expression = (201, 204)= . Purified human T cells do not upregulate CTLA4 following stimulation with anti-TCR mAb alone but the addition of either an immobilized form of soluble ICAM-1 or VCAM-1 resulted in significant expression of CTLA4 = (201, 204)= . It is, therefore, likely that several signaling pathways play an important role in controlling the expression of CTLA4. 1.5.1.2 Regulation of T cell activation by CD28 and CTLA4 The proliferative response of T cells isolated from CD28 -/- mice, or CD28 +/+ mice in the presence of CD28 antagonists is substantially reduced = (205)= . Therefore, the CD28 signal is thought to be essential for T cell proliferation. However, some reports have suggested that CD28-mediated signaling may also be important in the regulation of T cell survival = (206)= . Increasing the strength of the TCR-mediated signal increased early proliferative responses, but had no effect on sustained cell survival. In contrast, CD28 ligation was not required for early proliferative responses but dramatically influenced long-term T cell survival = (205, 207, 208)= . Increased cell survival after CD28 ligation was linked to up-regulation of bcl-xL = (209)= . Both bcl-2 and the high molecular weight form of bcl-x, bcl-xL, protect lymphocytes from apoptosis = (210-212)= . These results suggest that the unique role of CD28 signaling is not to costimulate the initial activation of naive T cells, but is, in fact, to sustain the late proliferative response and enhance long-term cell survival. CD28 engagement has been shown to enhance the production of various cytokines, including IL-1, IL-2, IL-4, IL-5, tumor necrosis factor (TNF), and IFN-g = (195, 213, 214)= , and may promote the differentiation of Th2 cells = (215-217)= . In the absence of CD28 signaling naive T cells are biased toward a Th1 phenotype and CD28 -/- mice have a reduced Th2-dependent antibody response = (218)= and impaired Th2 differentiation = (219)= . Although there have been some reports that CTLA4-Ig selectively blocks Th1 cells = (220, 221)= , the evidence points to a role for CD28 in the differentiation of the Th2, but not Th1, subset of T cells = (222)= . Although CD28 is clearly a costimulatory molecule, the function of CTLA4 was initially less well understood. The work of Linsley et al. suggested a functional cooperation between CTLA4 and CD28 on activated T cells, based on the synergistic effects of anti-CTLA4 and anti-CD28 mAbs in promoting T cell proliferation = (201)= . In contrast, Krummel et al. suggested that CD28 and CTLA4 have opposing effects on the response of T cells to stimulation = (223)= . Further evidence for a role for CTLA4 in the negative regulation of T cell activation was provided by the severe lymphoproliferative phenotype of CTLA4-deficient mice = (224-226)= . Allison and Krummel have proposed a model for explaining T cell regulation based on differential expression of CD28 and CTLA4, in which the outcome of T cell activation is determined by signals from the TCR, costimulatory signal from CD28 and inhibitory signals from CTLA4 = (227, 228)= . However, the role of CTLA4 in T cell activation is still controversial = (229)= . 1.5.1.3 B7-1 and B7-2 B7-1, a B cell activation molecule first described in 1981 by Yokochi et al. = (230)= , was the first ligand to be identified for CD28, and later for CTLA4 = (231)= . In addition to its expression on aB cells, B7-1 was also detected on a variety of APCs including dendritic cells = (232-235)= , Langerhans cells = (233, 236)= , activated monocytes = (232, 237, 238)= and activated T cells = (232, 233, 239-242)= . Both the human and mouse B7-1 genes are members of the immunoglobulin superfamily = (243, 244)= . The functional importance of the B7-1 molecule has been demonstrated in a number of studies. Both anti-CD3 mAb and PMA-induced T cell proliferation was augmented by the addition of B7-1 transfectants. Proliferation was shown to be enhanced in a CD28-dependent fashion since T cell activation was blocked by anti-CD28 mAb = (245, 246)= . Furthermore, the potent costimulatory role of B7-1 was demonstrated in vivo in transgenic mice in which B7-1 was ectopically expressed on pancreatic b cells = (234, 247, 248)= . Despite the apparent ability of B7-1 to provide sufficient costimulation when expressed on transfectants or in transgenic mice, it has been very difficult to demonstrate its function on normal APCs in mice. Anti-B7-1 mAbs minimally inhibit a primary mixed lymphocyte response (MLR), while CTLA4-Ig inhibits the response by as much as 80% = (234)= . Additionally, the staining of either LPS-activated B cells or whole spleen with CTLA4-Ig was not inhibited by anti-B7-1 mAbs, suggesting that an additional CTLA4 ligand existed = (234, 249-251)= . Finally, B7-1-deficient mice were shown to be capable of mounting an immune response to nominal antigens and APCs isolated from these mice could be stained with labelled CTLA4-Ig = (252)= . These observations led to the identification and eventual cloning of a second B7 family member, B7-2 = (232, 253)= . The human B7-2 gene was cloned by Freeman et al. = (253)= and Azuma et al. = (232)= independently. The mouse homologue of B7-2 has also been cloned = (254)= . These genes share only 25% amino acid homology with their B7-1 counterparts. Similar to B7-1 transfectants, B7-2 transfectants augment T cell proliferation and IL-2 production following suboptimal stimulation with anti-CD3 mAb or PMA. This costimulation was inhibited by either CTLA4-Ig or anti-CD28 mAb, but not by anti-B7-1 mAb, demonstrating therefore that B7-2 binds to both CD28 and CTLA4 = (232, 253, 255)= . While anti-B7-1 mAbs have been inefficient in their ability to block primary allogeneic MLRs, in most cases, the anti-B7-2 mAbs inhibited the responses to level similar to CTLA4-Ig = (239, 256, 257)= . A combination of both anti-B7-1 and anti-B7-2 mAbs were the most effective at inhibiting the MLR = (191, 258)= . The role for both molecules in primary responses is further supported by the finding that a combination of anti-B7-1 and anti-B7-2 mAbs can induce anergy = (191)= . 1.5.1.4 Different functional effects of B7-1 and B7-2 The expression of B7-1 and B7-2 varies on B cells = (233)= , T cells = (232, 233, 239-242)= , macrophages = (233)= , and dendritic cells = (232-235)= depending on the activation state of the cells. An additional level of complexity involves the ability of cytokines to either induce or suppress the expression of these costimulatory ligands. Therefore, either B7-1 or B7-2 may dominate during different stages of the immune response. Furthermore, there are an increasing number of studies to suggest that these two molecules do not bind the CD28 and CTLA4 molecules similarly = (259)= . Both B7-1 and B7-2 bind to CTLA4-Ig with a 20-100-fold higher avidity than to CD28-Ig. In addition, B7-1 was found to have a slightly higher avidity for CD28 and CTLA4 than B7-2. More significantly, B7-2 dissociated more rapidly from CTLA4-Ig than B7-1, and CTLA4-Ig was less effective at inhibiting B7-2-dependent responses = (259)= . Kuchroo et al. showed that T cells from a myelin basic protein-specific TCR transgenic mouse secreted predominantly IFN-g when cultured in the presence of anti-B7-2 mAbs, while cultures carried out in the presence of anti-B7-1 mAbs led to increased IL-4 secretion = (260)= . Evidence from Freeman et al. supported an important role for B7-2 in the signaling of IL-4 production. While transfectants of B7-1 or B7-2 induced similar levels of IL-2 and IFN-g, only B7-2 transfectants were able to induce IL-4. Continuous restimulation by the B7-2 transfectants resulted in the induction of greater amounts of IL-4 = (261)= . Taken together, it is implied that B7-1 and B7-2 may influence Th1 and Th2 development, respectively, but this is far from clear. 1.5.2 gp39/CD40 pathway CD40 is a 50-kDa membrane protein which is a member of the TNF-receptor family. Originally its expression was thought to be restricted to B cells, dendritic cells and basal epithelial cells = (262)= , but more recently it has been shown to be functionally expressed on macrophages = (263)= , Langerhans cells = (264)= , endothelial cells = (265, 266)= , and thymic epithelial cells = (267)= . The ligand for CD40 is gp39, a type II, member of TNF family which is preferentially expressed on activated T cells and mast cells = (268)= . A number of lines of investigation have led to the conclusion that interaction between gp39 and CD40 are critical for the development of humoral immunity = (268-271)= . Loss of gp39 function resulted in a near total loss of thymus-dependent primary and secondary humoral immune responses and the development of B cell memory. Using mice whose gp39 or CD40 gene was disrupted by homologous recombination, three reports have recently established that gp39 is required for protective cell-mediated immunity to Leishmania major = (272, 273)= and to Leishmania amazonensis = (274)= . All three studies strongly suggested that in the absence of CD40 signaling, T cells were polarized towards a T helper 2-type response. Further evidence in support of this scenario was provided by the results of anti-gp39 mAb administration in a model of inflammatory bowel disease (IBD) = (275)= . Rectal instillation of an inflammatory hapten induces IBD, the hallmarks of which are elevated Th1 cytokines, IL-12 production, and inflammatory pathology. Treatment with anti-gp39 mAb prevented the inflammatory response and promoted the priming of Th2-type cells. CD40 also plays a direct role in the microbicidal activity of macrophages = (276)= . Two studies showed that gp39 triggers the production of nitric oxide (NO) = (277, 278)= . gp39 regulation of inducible NO production has also been observed in the context of allogeneic organ transplant, whereby anti-gp39 mAb allowed the long-term survival of cardiac transplants and this long-term survival was correlated with the block by anti-gp39 mAb of inducible NO synthesis = (279)= . Therefore, gp39 appears to be a multipurpose macrophage activator involved in the up-regulation of microbicidal activity, antigen presentation, and costimulation, as well as the induction of proinflammatory and inflammatory cytokines. The role that CD40 plays in the regulation of APC activity is best exemplified by studies using B cells as APCs. A reciprocal dialogue between inducible molecules both on T cells and APCs provides regulated activation of T cells, which is important for amplification of the in vivo immune response. The current understanding of T-cell activation is that naive T cells require two distinct signals for activation (Figure 1-2) = (280)= . The first signal is provided by the engagement of the TCR with MHC/peptide ligand on the surface of the APC, thus ensuring antigenic specificity. The second signal is delivered to T cells by costimulatory molecules such as B7-1 and B7-2 on the surface of APC via their interaction with CD28/CTLA4 on the surface of T cells. However, APCs do not normally express the necessary costimulatory molecules unless they are first activated. Recognition of the MHC/peptide complex by the TCR leads to the induction of gp39 on T cells, whereas simultaneous engagement of MHC/peptide by the TCR, and CD40 by gp39 leads to the upregulation of B7-1 and B7-2 on B cells (Figure 1-3-a) = (270, 271)= . This costimulatory activity induced on B cells then acts to amplify the response of the T cells. Several pieces of evidences support the idea that gp39/CD40 interactions are required for the induction of B cell-mediated costimulatory activity for T cell activation: (i) mAbs against gp39 or CD40 inhibit activated T cells from inducing B7-1 and B7-2 on B cells = (281, 282)= , (ii) soluble gp39 induces costimulatory activity on B cells = (283)= . An alternative role of CD40/gp39 might be independent of B7/CD28 pathway as a direct T cell costimulator (Figure 1-3-b). Cayabyab et al. have demonstrated that P815 cells transfected with CD40 were able to induce T cell proliferation. Moreover, this proliferation induced by CD40-transfected cells could not be prevented by anti-B7 mAb = (284)= . van Essen et al. have demonstrated that administration of recombinant CD40 in vivo to CD40 -/- mice restored the helper function of T cells = (285)= . Therefore, a signal from gp39 to CD40 is known to be important in B cell activation, isotype switching and germinal centre formation, while the reverse signal (i.e. from CD40 to gp39) may be involved in T cell activation. 1.6 Protocols to induce tolerance to alloantigens 1.6.1 Acquired tolerance The induction of specific unresponsiveness by the administration of antigen from the organ donor before transplantation was first demonstrated to be a mechanism for inducing tolerance to alloantigen in vivo by Medawar et al. = (12)= . They administered semiallogeneic bone marrow and spleen cells to newborn mice and thus took advantage of the immaturity of the neonatal immune system. They were able to achieve long-term survival of skin allografts when these mice were transplanted as adults without the need for further immunosuppression. However, such approaches were believed to be limited to the foetal or very early neonatal period. 1.6.2 The blood transfusion effect (antigen induced immune hyporesponsiveness in transplantation) Reduced responsiveness to alloantigens can be achieved in adult recipients by preoperative blood transfusion. Clinically, this beneficial effect was shown by Dossetor et al. in 1967 = (186)= and then Morris et al. = (187)= . Before these studies, dialysis patients were expected to be more difficult to transplant because they had been repeatedly transfused and it was anticipated that they might have become sensitized. The powerful influence of blood transfusion on graft survival was confirmed by several prospective studies = (188, 286)= and similar results have been obtained in the rodent = (189, 190)= , the dog = (287, 288)= , and non-human primates = (289)= . Several investigators have studied the enhancing potential of red cells = (290)= , platelets = (291)= , B cells = (292, 293)= and lymphocytes = (294, 295)= . In a rat kidney model, Wood et al. showed that purified erythrocytes induced prolonged allograft survival and demonstrated that this effect was donor-specific and only effective if given by the i.v. route = (290)= . They concluded that MHC class I antigen could induce specific suppression of rejection of a renal allograft because erythrocytes express MHC class I antigens, not but MHC class II antigens. Superina et al. showed that pretreatment of C3H mice (H2k) with L cells expressing the donor-specific H2-Db class I molecule (L-Db cells), resulted in a slight prolongation of C57BL/10 (H2b) hearts transplanted 14 days later = (296)= . This studies were extended by experiments in which recipients were pretreated with L cells expressing H2-Db, H2-Kb MHC class I antigens or H2-IAb MHC class II antigens either alone, or in combination. 40% of the recipients treated with cells expressing only H2-Kb donor antigens accepted their grafts = (297)= . Although these protocols could induce donor-specific prolongation of graft survival, all grafts were eventually rejected in the majority of studies. 1.6.3 The use of mAbs to modulate immune responsiveness in the adult The rejection response is initiated by T cells. It is therefore a logical progression to attempt to inhibit graft destruction by the use of antibodies specific for T cells. Polyclonal preparations of antilymphocyte or antithymocyte globulins have been used as immunosuppressive agents for many years = (298, 299)= . However, it is difficult to produce polyclonal reagents that have consistent reactivity and potency. Furthermore, as understanding of the cell surface molecules expressed by leukocytes has increased, it has become clear that there may be advantages to targeting distinct subpopulations of T cells. mAbs may provide the solution to both of these problems. A mAb is produced by a single B cell clone that has been immortalized and that will theoretically produce antibodies with a single defined antigen specificity = (299)= . This approach has yielded encouraging results in experimental models with donor-specific unresponsiveness induced by the administration of mAbs targeting the CD4 = (300)= , LFA-1/ICAM-1 = (113)= , IL-2 receptor = (301)= , the B7 family/CD28 interaction = (192)= , and the gp39/CD40 interaction = (279)= . 1.6.3.1 Anti-CD4 mAb CD4 is expressed by MHC class II-restricted T cells = (302)= . It interacts with the b2 domain of the MHC class II molecule present on the APCs during antigen recognition through the TCR = (303, 304)= . Depletion of the CD4+ T cell subset was first shown to result in skin allograft (full MHC mismatch) prolongation by Cobbold et al. in 1984 = (305)= . Subsequently peri-operative treatment with anti-CD4 mAb has been shown to induce the prolonged survival of renal = (306, 307)= , cardiac = (308, 309)= , islet = (310)= and skin allografts = (311, 312)= . The efficacy of a particular anti-CD4 mAb as an immunosuppressive agent in vivo is dependent on its molecular properties, including epitope specificity, affinity and framework region characteristics = (313)= . First, the effect of anti-CD4 mAb was thought to be dependent on its ability to deplete CD4+ cells. However, in a mouse model, non-depleting anti-CD4 mAb also induced indefinite prolongation of fully allogeneic cardiac grafts = (314, 315)= , suggesting that elimination of CD4+ cells was not essential to induce graft prolongation. In some studies using CD4 mAb to induce tolerance, tolerance has been shown to result in a switch from a Th1 to a Th2 response = (125)= . 1.6.3.2 Anti-LFA-1 (CD18/CD11a) mAb LFA-1 is a member of the integrin family and is involved in leukocyte adhesion by binding to members of the ICAM family = (316)= . LFA-1 is involved in the antigen-independent adherence of leukocytes to endothelial cells, an important event when recipient leukocytes infiltrate the graft after transplantation, as well as the antigen-dependent interaction of leukocytes with APCs = (317)= and stimulation after antigen recognition by TCR = (318, 319)= . Monoclonal antibodies specific for LFA-1 have been used in transplantation studies and have been shown to induce prolonged survival of grafts in rodent 抜けてるよ!! 1.6.3.2 Anti-LFA-1 mAb 1.6.3.3 Anti-IL-2 receptor mAb 1.6.3.4 Anti-B7 mAb and CTLA4-Ig 1.6.3.5 Anti-gp39 mAb 1.6.4 Tolerance induction prior to transplantation 1.6.4.1 Anti-CD4 mAb + alloantigen 1.6.4.2 CTLA4-Ig + alloantigen 1.6.4.3 Anti-gp39 + B cells Chapter 2: Materials and methods 2.1 Animals 2.1.1 Mice C3H/He (H2k) (abbreviated as C3H), CBA.Ca (H2k) (abbreviated as CBA) and C57BL/10 (H2b) mice were purchased from Harlan Ltd. (Bicester, Oxfordshire, U.K.). CBK mice, a MHC class I, Kb, transgenic strain (H2k+Kb), were the kind gift of Professor A.L.Mellor = (343)= . All mice were housed in conventional facilities of the Biomedical Services Unit, John Radcliffe Hospital, Oxford. Some studies took place at the Emory University in Atlanta. In these studies, CBA, C57BL/6 (H2b), and F1(C57BL/6 x 129) (H2b) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, U.S.A.). CD40 knockout (-/-) (H2b) and gp39 -/- (H2b) mice were kindly provided from Dr. Raif Geha of Boston Childrens Hospital and Dr. Elaine Thomas at the Immunex Corp. in Seattle. F1(C57BL/6 x 129) (H2b) mice were used as wild type control of these knockout mice. All mice were used between 8 and 12 week old in accordance with the Animals (Scientific Procedures) Act 1986. A list of mouse strains used in this study, together with their H2 genotypes is shown in Table 2.1.
2.1.1 Rats Lewis rats (6 to 8 week old) were used to prepare Concanavalin A (Con A) supernatant. (LOU x DA) F1 rats were used to grow hybridomas for the production of mAbs from Ascites. They were bred and housed in the Animal Facility of the Department of Human Anatomy, University of Oxford. 2.2 Tissue culture media and reagents for cell preparation Phosphate buffered saline (PBS) Heat inactivated Foetal Calf Serum (FCS)
L-glutamine Antibiotics 2-mercapto-ethanol (2ME) RPMI 1640 (Roswell Park Memorial Institute 1640 medium) Complete RPMI tissue culture medium (R10) DMEM (Dulbecco's modified Eagle's medium) Complete DMEM PBS/EDTA
Tris-buffered ammonium chloride (TBAC) TBAC was used for the lysis of erythrocytes. 90ml of 0.16M NH4Cl was added 10ml of 0.17M Tris base and adjusted pH to 7.2 using 1M HCl. Sephadex G-10 Percoll "Dense" bovine serum albumin (BSA) Dimethyl sulphoxide (DMSO) Phorbol myristate acetate (PMA) Ionomycin 3H-thymidine Chromium 51 (51Cr) Sodium azide (NaN3) Triton-X 100 solution Freezing medium 2.3 Cell lines 2.3.1 RMA and P815 The mouse T lymphoma cell line RMA (H2b) and the mastocytoma cell line P815 (H2d) were used as targets in the cytotoxicity assay. The RMA and P815 cell lines were maintained at a density between 5x 104 to 5x 105 cells/ml in complete DMEM at 37 oC in a humidified CO2 air incubator. 2.3.2 L-Kb cells Fibroblasts (L cells) from C3H (H2k) mice were previously stably transfected by Dr. Joren C. Madsen in this laboratory with the MHC class I genes coding for the a chain of the K (L-Kb cells) loci derived from mice with an H2b genetic background, and kindly provided for this study. Untransfected L cells and L-Kb cells were grown in complete DMEM. After few days, PBS/EDTA was used to remove adherent cells from tissue culture flasks. Cells were washed and subdivided into new culture flasks for the maintenance of cell growth during the study. 2.4 Stock cells Cell lines were stored at -196 oC, under liquid nitrogen. The procedures used for freezing and thawing of cells is described below. 2.4.1 Freezing Cells were frozen when in growth phase, usually one or two days after passage. Cells were counted, and centrifuged at 1400 rpm, and 4 oC, for 10 minutes. The supernatant medium was discarded, and the cells were resuspended to between 1x and 5x 106 cells/ml in freezing medium (10% DMSO in R10). 1ml of the cell suspension was then dispensed into each sterile, ice-cold, freezing ampoule. The ampoules of cells were immediately packed with cotton wool in a box and transferred to an automated controlled rate freezer (-80 oC). One day later, the ampoules were transferred to liquid nitrogen, for storage. 2.4.2 Thawing Frozen ampoules of cells were taken directly from liquid nitrogen to a water bath at 37 oC, in order to thaw the cells as quickly as possible. Cells were transferred to a 15ml tube and 1ml of R10 added and mixed well, and this procedure repeated up to 15 ml of volume. Depending on the number and viability of the cells, they were either placed into flask, or into 2ml or 5ml wells, at a final concentration of approximately 2x to 5x 105 cells/ml. 2.5 Preparation of specific cell populations 2.5.1 Preparation of splenocytes A single cell suspension was prepared by gentle dissociation of a mouse spleen through a sterile stainless sieve. The leukocytes were overlaid on to a Ficoll gradient (Histopaque-1083, 6.41g/dl, Sigma-Aldrich). The cells at the interface were harvested after centrifugation at 2000 rpm for 20 minutes and washed twice before use. 2.5.2 Preparation of B cells: Resting B cells were prepared by a modification of Defranco et al. = (346)= . Cell preparation was performed in PBS supplemented with 10% heat-inactivated FCS (PBS/FCS). Splenocytes were depleted of erythrocytes by incubation with TBAC for 5 minutes at room temperature. This reaction was stopped by adding PBS/FCS at over 10 times of the TBAC volume. Next, T cells were depleted by antibodies and complement lysis. Antibodies to CD4 (YTS191) and CD8 (YTS169) (the hybridomas were kindly provided by Professor H Waldmann, Oxford, U.K.) were added to the cells for 30 minutes on ice. After washing twice the cells were incubated with rabbit serum as a source of complement (Oxford Transplant Centre, Churchill Hospital, Oxford, U.K.) for 45 minutes at 37 oC. In order to deplete macrophages and dendritic cells, the remaining cells were passed over two sequential Sephadex G-10 columns. Finally, the eluted cells were centrifuged over a discontinuous Percoll gradient to collect low density cell population. Percoll discontinuous gradient was prepared by overlaying 4ml of 70%, 60% and 50% Percoll from the bottom of 15ml tube. The cell suspension was overlaid on the top of the Percoll discontinuous gradient and centrifuged at 2600 rpm for 20 minutes at room temperature without braking of the centrifuge machine. Cells at the 60 to 70% interface were collected and used as resting B (rB) cells. A typical preparation of rB cells as analysed by flow cytometry was found to be 95.7% MHC class II+, 1.2% B7-1+, 2.5% B7-2+ , 5% CD3+ and 92.3% sIg+. For preparation of LPS activated B (aB) cells, the resting population (1x 106 cells/ml) was cultured with LPS (30mg/ml) for 2 days in R10 at 37 oC in a humidified CO2 air incubator. In all cases, cells were washed 3 times before use. A typical preparation of aB cells as analysed by flow cytometry was found to be 96.1% MHC class II+, 49.5% B7-1+, 77.7% B7-2+, <2% CD3+ and 96.9% sIg+. 2.5.3 Preparation of Low Density Adherent Cells (LODACs) LODACs were prepared as described by Steinman et al. = (344, 347)= . A single cell suspension was made from mouse spleens. After centrifugation, the supernatant was discarded and the pellet was resuspended with "dense" BSA at over 10 times the volume of pellet. After spinning at 2600 rpm for 20 minutes at 4 oC without braking, the interface was collected and washed twice with R10. The cells were layered onto a plastic dish (1x 108 cells/dish, Falcon 3003, Lincoln Park, NJ, U.S.A.) in 10ml of R10. After incubating the plate for 90 minutes at 37 oC, nonadherent cells were removed, the plates rinsed and the medium replaced. After incubation for another 16 hours at 37 oC in a humidified CO2 air incubator, nonadherent cells were collected and used as LODACs after washing. A typical preparation as analysed by flow cytometry was found to be 70% N418+, 92.2% MHC class II+, 68.8% B7-1+, 88.2% B7-2+ and 9.4% sIg+. Therefore, LODACs were used as a dendritic cell enriched population. 2.5.4 Preparation of nylon wool purified T cells Splenocytes were loaded onto a nylon wool column (Robbins Scientific, Sunnyvale, CA, U.S.A.) (6g nylon wool was packed into a 40ml column). After incubation for over 1 hour at 37 oC in a humidified CO2 air incubator, cells were eluted slowly in a humidified CO2 air incubator using R10 and collected. They were depleted of erythrocytes with TBAC and used as T cells after washing. Typical preparations analysed by FACS was found to be approximately >80% CD3+, <10% IA+ and <10% Ig+. 2.5.5 Preparation of FACS sorted T cells Nylon wool passed T cells were made from spleen of C57BL/10 mice. These cells were stained with FITC-conjugated anti-IA(b, d) mAb for 30 minutes on ice after washing twice. The cells were sorted negatively by FACS Vantage with turbo sort option (Becton Dickinson, Mountain View, CA, U.S.A.). Typical preparations as analysed by FACS was found to be >90% CD3+, <0.5% IA+ and <1% Ig+. 2.6 Flow cytometry 2.6.1 Normal staining 25オl of viable cells (1x 107 cells/ml) were incubated for 30 minutes on ice with primary antibodies in PBS supplemented with 2% FCS and 0.02% sodium azide. When nonconjugated rat mAbs were used as the first stage, cells were washed twice before being incubated for a further 30 minutes on ice with either FITC or PE-labelled goat anti-rat immunoglobulin reagent (Sigma-Aldrich) blocked with 10% mouse serum. When biotinylated-antibody was used in the first incubation, streptavidin-phycoerythrin (Southern Biotechnology Associates, Birmingham, AL, U.S.A.) or streptavidin-Cy-chrome (Pharmingen Ltd., San Diego, CA, U.S.A.) was used for the second incubation. When nonconjugated hamster mAbs were used as the first stage, cells were incubated for a further 30 minutes on ice with biotinylated-anti-hamster antibody followed by streptavidin-phycoerythrin. After two more washes, the fluorescence intensities of 10,000 cells were determined using a FACSort (Becton Dickinson) flow cytometer. Samples were generally analyzed immediately after staining. Where necessary samples were stored in 1% formaldehyde/PBS at 4 oC in the dark for up to 5 days. Table 2.2 shows the antibodies used in these studies. 2.6.2 Staining of B7-2 on IAb+ cells Single cell suspensions from the spleens of CBA mice pretreated with 1x 107 rB cells in the presence or absence of MR1 or LPS activated B cells from C57BL/10 were prepared 24 hours after i.v. injection. Resting B cells immediately after isolation were used as a negative control of B7-2 expression. IAb+ cells were labelled with biotinylated-B21.2 mAb and streptavidin-phycoerythrin. B7-2 expression on these cells was determined by staining with FITC conjugated anti-B7-2 mAb. IAb positive cells were gated and 5000 counted for analysis. Table 2.2 shows the antibodies used in this study. 2.6.3 Staining cells for tracing in vivo Carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes, Leiden, The Netherlands) was used for tracing cells after i.v. injection. 5mM stock solution was stored at -80 oC until required. Single cell suspension for injecting in vivo was incubated with CFSE for 10 minutes in a 37 oC waterbath with regular agitation. 1オM CFSE (1/5000 of stock solution) was the optimal when cells were detected one day after i.v. injection. The reaction was stopped by placing the tubes on ice and washing immediately. The cells were washed 3 times with ice-cold RPMI 1640 for in vivo use. The antibodies used for FACS analysis in this study were as follows:
2.7 Allo-antibody assay For characterization of recipient sera, 4x 105 L-Kb cells were incubated with the recipient sera for 45 minutes on ice. Cells were washed three times with PBS containing 2% (v/v) FCS and 0.02% (v/v) sodium azide, then incubated for an additional 45 minutes with FITC-labelled goat anti-mouse IgG (1:100 dilution; GAM-FITC, g chain specific; Sigma-Aldrich). After two further washes with PBS-FCS-azide, the cells were resuspended in 200ml 2% (v/v) formaldehyde and 200ml PBS-FCS-azide. Sera from naive untreated mice were used as the negative control in all experiments and tissue culture supernatant of TIB139 (anti-Kb mAb) was used as the positive controls. The FACS assays were run on a FACSort (Becton Dickinson). 2.8 Functional assays in vitro 2.8.1 Mixed leukocyte culture (MLC) T cells prepared by nylon wool purification were used as responders (2.5x 105 cells per well). Stimulator cells were irradiated with 3000 rad before use. Cells were cultured in 200オl of R10 in triplicate (96-well U-bottom plates, Life Technologies) for 4 days. 3H-thymidine was added for the last 16 hours of the incubation. Cultures were harvested onto glassfibre filter mats (Wallac, Turku, Finland) and counted for thymidine incorporation using a Betaplate counter (Wallac). In second MLC, 3 days was chosen as culture period instead of 4 days. 2.8.2 Labelling 51Chromium Target cells were counted and spun down. These cells were resuspended with 100オCi 51Chromium and cultured for 60 minutes at 37 oC. After washing three times, they were resuspended into at the desired concentration and used as target cells. 2.8.3 Cytotoxic T lymphocyte (CTL) assay CTL were assayed using a 51Cr release assay. In order to examine the ability of the fractionated cells to generate cytotoxic activity, CBA (H2k) or C3H (H2k) splenocytes as effector cells were cultured with irradiated allogeneic C57BL/10 (H2b) stimulators, either LODACs, aB cells or rB cells for 4 days. Effector cells were added in triplicate to 5000 51Cr-labelled RMA (H2b) and P815 (H2d) as specific and non-specific targets, respectively, at different effector/target ratios in a total volume of 200オl of R10. Targets were also incubated with medium alone or 1% Triton X-100 detergent as spontaneous and maximum lysis control, respectively. Plates were incubated for 4 hours at 37 oC in 5% CO2. 20オl of the cell-free supernatant was then removed carefully from each of the wells onto a spot-on filter mat (Wallac) and assayed in a Betaplate counter. Mean values were calculated from replicate wells, and the results were expressed as percent specific lysis according to the formula; [(experimental counts - spontaneous counts) / (maximum counts - spontaneous counts)] x100. Also, this experiment was performed to examine the ability of splenocytes from mice accepting cardiac grafts for long-term to generate cytotoxic T cells for specific and non-specific targets. In this case, splenocytes from recipient mice were used as effectors. 2.8.4 Limiting dilution analysis (LDA) For the analysis of allospecific CTLp frequencies, LDA was performed. Briefly, graded numbers (from 100,000 to 1562 cells per well in twofold dilutions) of single cell suspensions of fresh splenocytes were cultured in 24 replicates (96-well U bottom plates, Life Technologies) with 100,000 irradiated (3000 rad) stimulator cells (splenocytes) in a total volume of 200オl of R10 contained 15% Con A supernatant as a source of IL-2. After 7 days of culture each well was tested individually for cytolytic activity against 5000 51Cr labelled-target cells, as described above. Microcultures were considered cytolytic if the observed chromium release exceeded the spontaneous release (stimulators only) plus 3 standard deviations. All frequency determinations of CTLp were consistent with a single hit kinetic model, indicating that only one cell type (i.e. the cytotoxic T cell was limiting). 2.9 ELISA (enzyme-linked immunosorbent assays) 2.9.1 Reagents for ELISA Tween-20: Polyoxyethylene-sorbitan monolaurate (Sigma-Aldrich)
2.9.2 Methods of ELISA
Day 1
Day 2
Day 3 (Avidin-peroxidase) (Substrate) 2.9.3 mAbs for ELISA Table 2.3 shows the antibodies used in ELISA study.
2.10 Monoclonal antibody purification 2.10.1 Production of mAb in tissue culture supernatant The hybridomas of YTS 191 and YTS 169 were kind gifts from Professor H. Waldmann, Oxford, U.K.. These hybridomas were expanded at a constant concentration of 2x 105 cells/ml until the desired volume of supernatant had been reached. The cultures were allowed to grow to exhaustion which was defined by less than 80% cell viability, and usually required 5 to 6 days. The supernatant was centrifuged (1200 rpm for 7 minutes) prior to filtering through a 22オm filter (Cat No. GSWP 04700, Millipore Ltd. Watford, U.K.). It was stored in 1 to 3ml aliquots at -80 oC prior to use in immunological assays or purifying B cells.
2.10.2 Production of mAb from rat ascites injected with hybridoma Antibodies, YTS 169 and YTS 191.1, were used for purifying B cells in this study. (1) Production of ascites Adult (LOU x DA)F1 rats were housed at the department of Anatomy, University of Oxford animal care facility. The rats were pristane primed under ether general anaesthesia by injecting 0.25ml of neat pristane (2,6,10,14-tetramethylpentadecane, Koch-Light Ltd., Haverhill, U.K.) into each of the four quadrants of the peritoneal cavity. Ten days after the pristane injection 2ml of hybridomas at 1x 107 cells/ml were injected into each quadrant. The rats were observed daily for ascites production. 10 to 14 days after injection of cells, when ascites accumulation was judged to be maximal, and before the rats became moribund, they were killed using ether, the peritoneal cavity opened via a midline incision, and the ascites aspirated. The ascites was centrifuged at 1600 rpm at 4 oC for 10 minutes to remove cellular debris and then stored at -80 oC. (2) Ammonium sulphate precipitation Ascites was removed from the freezer and allowed to thaw at room temperature. It was centrifuged at 1600 rpm at 4 oC for 10 minutes. Purification using 50% saturation with ammonium sulphate was then carried out. This was done by slowly adding 29.5g of (NH4)2SO4 per 100ml volume of ascites while mixing slowly on a magnetic stirrer. Stirring was continued at 4 oC for 30 minutes. This solution was then spun in the ultracentrifuge (L8-80M, Beckman, U.S.A.) at 20,000 rpm for 30 minutes at 4 oC and the supernatant discarded. The pellet was resuspended in distilled H2O at 4 oC at half the original volume. Dialysis of the antibody using a dialysis tube (cut off molecular weight 12,000-14,000) (Medicell international Ltd., London, U.K.) was performed overnight (2 changes) in 25mM Tris Buffer pH 7.4. (3) Ion exchange chromatography (DEAE) The DEAE (diethylaminoethyl)-Sephacel (Pharmacia) ion-exchange column was constructed in a 30ml syringe barrel and equilibrated with at least 20 volumes of 25mM Tris pH 7.4 buffer. After overnight dialysis the ammonium sulphate purified ascites was loaded onto the column and washed through the column with 25mM Tris pH 7.4 buffer until the OD at 280nm was zero by a spectrophotometer ( DU-68, Beckman). Continuous gradients from 0 to 150mM NaCl with 25mM Tris pH 7.4 buffer (300ml of 25mM Tris pH 7.4 without NaCl and 300ml of 25mM Tris pH 7.4/300mM NaCl) was used for collecting the peaks of protein. All fraction (6ml/each) were collected and pooled according to peaks of OD at 280nm. The protein content was estimated by measuring the OD at 280nm. Total protein (mg/ml) = OD (280nm)/1.4 All pools for use were analysed by SDS-PAGE. For purity, the pool selected for use was then dialysed into PBS, aliquoted in vials and stored at -80 oC until required. (4) Determination of Purity: SDS-polyacrylamide gel electrophoresis (PAGE) Reagents for SDS-PAGE Solution A: 500ml of 30% (w/v) acrylamide (BDH Ltd., U.K.) and 0.8% (w/v) N,N'-methylene-bis acrylamide (Sigma-Aldrich) was made by adding 10.6g of the former and 6.6g the latter to 500ml of distilled water and then filtered. Solution B: 500ml of 0.75M Tris HCl pH 8.8 was made by adding 45.4g of Trizma Base (Sigma-Aldrich) to 500ml of distilled water and adjusting the pH to 8.8 with concentrated HCl. Solution C: 500ml of 1M Tris HCl pH 6.8 was made by adding 60.5g of Trizma Base to 500ml of distilled water and adjusting the pH to 6.8 with concentrated HCl. Solution D: 100ml of 10% (w/v) sodium dodecyl sulphate (SDS) was made by adding 10g of lauryl sulphate (Sigma-Aldrich) to 100ml of distilled water. Solution E: 20ml of 0.1% (w/v) ammonium persulphate was made by adding 0.02g of ammonium persulphate (Bio Rad) to 20ml of distilled water. (This solution must be made fresh, the others may be made as stock solutions.) Solution F: N,N,N',N'-tetramethyl ethylenediamine (TEMED) (Sigma-Aldrich) Running Buffer: 500ml of 10x stock solution is made by adding 15.5g of Trizma Base and 72g of glycine (Sigma-Aldrich) to 500ml of distilled water and adjusting the pH to 8.3 with concentrated HCl. To use the stock solution was diluted 1/10 with distilled water and 0.1% of SDS (lauryl sulphate) added. Sample buffer: 100ml of 0.2M Tris 8.0M Urea 2% SDS pH 8.0 sample buffer was made by adding 2.4g of Trizma Base, 48.05g of Urea (BDH) and 2.0g of SDS (lauryl sulphate) to 100ml of distilled water. Bromophenol blue (BPB): was used for the marker to indicate the position of fast running protein in a gel. BPB was made by adding one microspatula of BPB to 10ml of sample buffer. Dithiothreitol (DTT): was used for reducing S-S bonds of protein. 7.75mg of DTT (Sigma-Aldrich) was added in 1.25ml of sample buffer. This is a 4x concentrated solution. Iodoacetamide (IAA): was used to stabilize the proteins reduced with DTT. 37mg of IAA (Sigma-Aldrich) and 30オl of bromophenol blue was added to 5ml of sample buffer. This is a 2x concentrated solution. Coomassie blue staining solution: 1g of Coomassie brilliant blue R (Sigma-Aldrich) was added to 73.6ml of acetic acid, 364ml of methanol and 364ml of distilled water. This solution was filtered prior to each use. Destaining solution: This solution was made by adding 150ml of acetic acid to 100ml of methanol and 1750ml of distilled water. Casting of the gel The components of the gel slab casting apparatus (Mighty smallェ, Hoefer Scientific Inst., San Fancisco, CA, U.S.A.) were cleaned with 70% ethanol and dried with acetone prior to assembly. The gel was mixed from the solutions listed above as follows:
The gel cassette was filled, and the gel covered with distilled water to prevent air contact while it set. After the gel was set (approximately 30 minutes) the distilled water was removed with a paper towel and replaced with the stacking gel. The sample well former comb was placed in the stacking gel. Preparation of the sample The aim was to apply 20オg of protein to each sample well. The maximum volume that was added to each well was 10オl. The volume of sample was therefore calculated by: Running of the gel The comb was removed from the stacking gel and the plate was inserted into the gel tank. The running buffer was loaded into the tank and a vigorous effort made to rid the chamber of air bubbles. Using a Hamilton syringe each sample and the molecular weight markers were added to the sample wells. Constant current was applied to the gel at 20mA/gel on a power supply (2103 Power supply, LKB Biochrom, U.K.) until the BPB was seen near the bottom of the gel; this took approximately 1 hour. Staining the gel Coomassie blue was used to visualise the final position of proteins in the gel. The gel was incubated (for between 1 hour to overnight, depending on convenience) in the coomassie blue stain at room temperature, and then destained by incubation in frequently changed solution of destain until a clear background was obtained. Reading the gel A gel was sealed with plastic sheets using Sealing Machine (Hulme Martin, London, U.K.). Bands were read and analyzed with Bio Image Whole Band Analyzer (Millipore, MI, U.S.A.). 2.11 Paraffin histology 2.11.1 Hematoxylin and eosin staining Morphologic analysis of cardiac grafts was done on tissue prepared by paraffin wax section processing followed by hematoxylin and eosin staining which was performed by Dr. David Hughes, in this department. 2.11.2 Masson trichrome staining To examine connective tissue components, masson trichrome staining was performed by Dr. David Hughes, in this department. 2.12 Monoclonal antibodies for in vivo use 2.12.1. Anti-gp39 mAb (MR1) MR1 is a hamster mAb specific for a counterreceptor of CD40, gp39. It was kindly provided by Drs. Alejandro Aruffo and Diane Hollenbaugh (Bristol-Myers Squibb Pharmaceutical Research Institute, Seattle, WA, U.S.A.) = (266)= . 2.12.2 CTLA4-Ig CTLA4-Ig is a human immunoglobulin Fc fusion of the extracellular domain of human CTLA4. It was kindly provided by Dr Peter S. Linsley (Bristol-Myers Squibb Pharmaceutical Research Institute) = (351)= . 2.13 Animal related techniques. 2.13.1 Anaesthesia Hypnormィ (Jensen Pharmaceutical Ltd., Oxford, U.K.) contains fentanyl citrate and fluanisone. The Hypnormィ stock solution was diluted with sterile saline to a working solution containing 10オg/ml fentanyl and 330オg/ml fluanisone. Midazolam (Hypnovalィ, Roche Products Ltd., Welwyn Garden City, U.K.) was diluted from the stock solution with sterile saline to give a working solution of 5000オg/ml. Mice were injected subcutaneously with 0.3ml of both Hypnormィ and midazolam working solutions before procedure. Transplant recipients also received a local anaesthetic injected subcutaneously (along the midline) prior to the procedure. The local anaesthesia was provided by Marcaineィ 0.25% (bupivacaine hydrochloride BP, 2.64mg/ml, Astra, was diluted from a stock solution to a concentration of 0.8mg/ml with sterile saline (0.9%)) . 2.13.2 Blood collection Heparinized whole blood (100ml heparin/ml, Leo Laboratories Ltd., Princes Risborough, U.K.) was collected from anaesthetized C57BL/10 blood donors and stored on ice until required. Blood was used within 2 hours of collection. 2.13.3 Intravenous injection Intravenous injections were made through the penile or tail vein. For the penile vein injection, the mice were anaesthetized as described previously and placed in a supine position with the tail facing the operator. The penis was sterilized with 70% alcohol and stretched to find the penile vein in the middle line of dorsal penis. A 30 gauge needle was used to inject sample. On completion of the procedure, the recipient was placed under a warming lamp for 2-4 hours before being returned to conventional housing. For the tail vein injection, the mice were placed under warming lamp for a few minutes and put in a mouse holder. A 30 gauge needle was used to inject sample to the tail veins running along both sides of the tail. 2.13.4 Intraperitoneal injection The mouse was held by the operator's left hand. The neck was held with operator's thumb and index finger and the tail with the small finger and palm. The abdomen was sterilized with 70% alcohol and the intraperitoneal injection was made at the point below its navel with a 27 gauge needle facing its head. 2.13.5 Recipient pretreatment For skin grafts, recipient mice were pretreated with either donor 1x 107 rB cells, aB cells, unfractionated splenocytes, 5x 105 LODACs or 0.25ml DST intravenously 7 days before skin transplantation (Protocol-1, Figure 2-1). For cardiac grafts, recipient mice were pretreated with either donor 1x 107 rB cells, aB cells, unfractionated splenocytes, T cells, 5x 105 LODACs or 0.25ml DST, or 1x 107 CBA unfractionated splenocytes intravenously in the absence or presence of intraperitoneal single dose of 250オg MR1, 200オg CTLA4-Ig, or both 14 days before cardiac transplantation (Protocol-2, Figure 2-2). (Figure 2-1)
(Figure 2-2)
2.13.6 Skin grafting Recipient mice were anaesthetized as described previously, and shaved on the dorsal thorax. The skin was sterilized with 70% alcohol. A bed (approximately 16mm x 8mm) was prepared for the graft by carefully dissection of the recipients skin from the underlying fascia. The donor was killed by cervical dislocation, and the tail sterilized with alcohol. Full thickness skin from the tail of the donor was stripped from the base of the tail to its end, and was placed on a saline-moistened gauze. Individual grafts were prepared to fit the beds already prepared on the recipients = (352)= . Grafts were kept in place by a flexible colloid (Flexible collodion B.P. methylated, William Ramsom & Son, Herts, U.K.) and dressed by sterile gauze (povidone iodine non-adherent dressing, Inadineェ, Johnson & Johnson Medical, Ascot, U.K.) and covered with autoclave tape. The dressing was removed 7 days after the operation. The grafts were inspected daily until they were completely destroyed, at which time the grafts were considered rejected. 2.13.7 Heterotopic vascularized cardiac grafting (1) Introduction The mouse heterotopic fully vascularized cardiac transplantation model has come into widespread use since its introduction by Drs. Robert Corry and Paul S. Russell in 1973 = (353)= . This model is of particular value because it allows the fate of fully vascularized organ grafts to be evaluated in a species where a large number of genetically well-characterized strains and newer transgenic and gene knockout animals are available and for which there exists a very large number of immunological reagents. In this model the donor ascending aorta is anastomosed end-to-side to the recipient abdominal aorta and the donor pulmonary artery is sutured to the recipients inferior vena cava (IVC). Hearts transplanted heterotopically behave functionally as aorto-caval fistulae. Blood enters the donor ascending aorta from the recipient abdominal aorta and is diverted into the coronary arteries by the closed aortic valve. After the myocardium is perfused, venous blood drains into the right atrium through the coronary sinus and is pumped back into the recipients IVC by the right ventricle. The procedure was carried out using ideally an operating microscope (such as Carl Zeiss OPMI1-FC or OPMI6-CH, Germany) at a magnification of between 4x to 25x. Operating time was less than one hour and overall operative mortality was less than 5% in experienced hands. Details of the procedure are as follows: (2) Recipient preparation The recipient mouse was induced to urinate by placing gentle pressure on the lower abdomen before being anaesthetised. A midline abdominal incision was made from the pubic symphysis to the xyphoid. If the urinary bladder was still filled, it was emptied using a 25 gauge needle and 1ml syringe. A self-retaining retractor was placed to expose the abdominal contents. Using two sterile cotton buds the small intestine was gently deflected superiorly onto a piece of moistened sterile gauze on the thoracic cavity. Testes were deflected inferiorly in the same fashion when the recipient was a male. The testes and intestines were covered with the gauze, and are kept moist throughout the procedure with sterile saline. The meso-sigmoid was divided allowing a strip of moist gauze to pass under the sigmoid colon. The strip of gauze was now used to deflect the sigmoid colon to the animal's left side. Using two cotton buds the abdominal aorta and IVC were dissected free from the surrounding tissues in the retroperitoneum exposing the lumber arteries and veins which lay directly posterior coming from the great vessels in two or three groups between the renal vessels and the bifurcations inferiorly. The two or three groups of lumber vessels were ligated individually with 7-0 silk ties. If the recipient was male, two vessels on the IVC were found. These vessels were isolated from the IVC. A Scoville-Lewis clamp (Downs Surgical Ltd., U.K.) was positioned at the proximal side of bifurcation interrupting flow in both the aorta and IVC. Another clamp was positioned just below the renal vessels in a similar fashion. Clamping in this order ensured partially filled vessels which would facilitate the aortotomy and venotomy. If the IVC continued to fill after cross clamping, the ligation of lumber vessels should be checked. Under ideal conditions cross-clamping for as long as two hours was acceptable. (3) Donor operation The donor's abdominal cavity was opened with a large longitudinal incision. Abdominal contents were deflected to the left side of the animal exposing the IVC. Using a 1ml syringe and a 25 gauge needle, the IVC was canulated and as much blood as possible was aspirated. At this point, 1ml of cold heparin (diluted to 18,000U/ml final concentration with sterile saline) was administered via the IVC using another 1ml syringe and 25 gauge needle. The thoracic cavity was opened by cutting through the ribs along both sides of the thoracic spine all the way to the thoracic inlet. The anterior chest wall was deflected superiorly and held in place with a piece of adhesive tape. The IVC, azygous vein and the superior vena cava were ligated with 7-0 silk ties (Figure 2-3). The ascending aorta was cut below the brachiocephalic artery (Figure 2-3). The main pulmonary artery was cut proximal to its bifurcation (Figure 2-3). Anastomosis of the donor to recipient vessels was made considerably easier if the connective tissue between the ascending aorta and pulmonary artery was gently teased away at this stage. The pulmonary veins were ligated as a group with a single 7-0 silk tie (Figure 2.3). The donor heart was gently detached from the remaining connective tissues with blunt dissection and was placed in cold saline. (4) Implantation of the vascularised heterotopic heart graft The donor heart was placed into the recipients abdomen with the remnant of the ascending aorta lying towards the left side of the animal. An aortotomy was made in the abdominal aorta of the recipient using a 30 gauge needle. The aortotomy was then extended with micro-scissors superiorly to a length of equal to or slightly smaller than the opening of the donor ascending aorta. The donor ascending aorta was anastomosed to the recipient abdominal aorta using 10-0 Bear surgical sutureェ on a round-bodied 4mm (3/8) needle (Kyowa Precision Instruments Corp., Tokyo, Japan). A stay suture was placed at the proximal apex bringing the donor ascending aorta and the recipient abdominal aorta together (Figure 2-4-1). The ends of the stay suture were cut to a length of approximately 2mm and 5mm. Similarly, the distal apex of the donor aorta and the recipient aorta were brought together with a 10-0 suture and tied leaving a 5mm length of suture on one end and the needle on the other end. The anastomosis was then completed with the same suture in a counter-clockwise direction with a continuous running stitch (Figure 2-4-2). The distance from the distal apex to the proximal apex was covered with four stitches. The stitch was tied to the proximal stay suture. The second side of the anastomosis was completed in a similar manner continuing from the proximal apex to the distal apex and was tied to the distal stay suture (Figure 2-4-3). The donor pulmonary artery was now anastomosed to the recipient IVC. A venotomy was made in the same way as the aortotomy made at the level inferior to the aortotomy leaving a space between the two of approximately 2mm, which allowed easier access for anastomosing the second side (Figure 2-4-4). In contrast to the recipient's aorta anastomosis, a stay suture was first placed at the distal apex bringing the donor pulmonary artery and the recipient IVC together. The ends of the stay suture were tied leaving a 5mm length of suture on one end and the needle on the other end (Figure 2-4-5). Similarly, the proximal apex of the donor pulmonary artery and the recipient IVC were brought together with a 10-0 suture. The ends of the proximal stay suture were cut to a length of approximately 2mm and 5mm (Figure 2-4-6). Stay sutures in this order ensured the same length of both sides of donor pulmonary artery which made the anastomosis more straightforward. The anastomosis was then completed with the distal stay suture in a clockwise direction with a continuous running stitch. The distance from the distal apex to the proximal apex was covered with four throws (Figure 2-4-7). The stitch was tied to the proximal stay suture. At this point the needle was passed between the two donor vessels and the donor heart was flipped from the left side of the animal to the right (Figure 2-4-7). The second side of the anastomosis was completed in a similar manner continuing from the proximal apex to the distal apex and was tied to the distal stay suture. The anastomosis was performed similar to the aortic anastomosis except that the distal stay suture was done first and that a continuous running suture went in clockwise fashion (Figure 2-4-8). The distal clamp was removed first to check the anastomosis of the IVC. After approximate 5 to 10 seconds the vessel filled and the anastomosis became competent. Small pieces of moistened haemostatic agent Spongostan (Johnson and Johnson) were placed around each anastomosis. After ensuring adequate haemostasis the proximal clamp was slowly removed. Normally the donor heart filled with blood immediately, becoming bright red in colour, and initiating contraction of the heart. Provided that there was no further bleeding, the intestines and testes were returned to the abdominal cavity. The abdominal incision was closed in two layers with 4-0 chromic catgut with needle (Davis and Geck, U.K.). Separately, the muscle and skin layers were closed with a continuous running stitch. (5) Recovery On completion of the procedure, the recipient was placed on soft dry bedding material under a warming lamp for 2 to 4 hours with easy access to food and water. 2.13.8 Second cardiac allografts into the neck A second heart was transplanted into the recipient neck. The donor ascending aorta was anastomosed end-to-side to the recipient carotid artery and the donor pulmonary artery was sutured end-to-side to the recipient external jugular vein. 2.13.9 Assessment of graft survival Mice with non-functioning grafts within two days of transplantation were considered technical failures and were excluded. The function of transplanted hearts was followed by abdominal palpation
Absence of a palpable heart beat or electrocardiogram (ECG) activity was considered as rejection = (354)= . If a heart beat returned after an absence of more than 1 week, this was also counted as rejection. ECG was performed at day 50 and 100 days post transplant. Animals were anaesthetized as described previously and the electrodes were attached to record the ECG of both the transplanted and native heart. 2.13.10 Adult thymectomy The recipient was anaesthetized as described previously and placed in a supine position with the head facing the operator. The head was stretched and immobilized with a piece of 7-0 silk holding the upper incisor teeth of the animal to the operating board. A vertical incision about 1.5cm long was made in the neck from the midpoint of the chin to the level of the midclavicle. The thyroid glands were retracted and the muscles overlying the trachea were divided and splayed using 8-0 nylon stay sutures. Thymus was exposed by a median sternotomy to the second anterior rib. The left lobe of the thymus was gently retracted using forceps and the connective tissue dissected. This procedure was repeated until the left lobe was removed. The same technique was repeated to remove the right lobe. The skin was closed by a running stitch with a 4-0 cut gut suture. On completion of the procedure, the recipient was placed under a warming lamp for 2 hours before being returned to conventional housing. 2.13.11 Statistical analysis: Allograft survival between two groups was compared by Mann-Whitney U test corrected for ties using StatView SE + Graphic (Abacus Concepts Inc., U.S.A.) (*P<0.05, **P<0.01 and ***P<0.001 compared with naive control, unless otherwise mentioned). (Figure 2-3)
(Figure 2-4)
Chapter 3. Characterization of resting B cells in vitro 3.1 Introduction 3.1.1 Aim of this chapter The aim of this thesis was to investigate the ability of rB cells to induce tolerance to alloantigens in vivo. Therefore, first rB cells were purified and characterized in vitro. 3.1.2 Background Competent APCs such as dendritic cells, macrophages and activated B (aB) cells, which provide both signal 1 and signal 2, can activate T cells fully. In contrast, incompetent APCs, which can only provide signal 1, are thought to be unable to stimulate T cells and moreover may render T cells anergic (Figure 1-2) = (355)= . The incompetent APCs used in the majority of experiments (Section 1.3.4) do not exist under normal circumstances. This raises the question of what constitutes an incompetent APC under normal conditions. The most suitable candidate is thought to be a rB cell, which expresses MHC class II constitutively but does not express members of the B7 family = (235, 237, 356)= . Several experiments have shown that rB cells can act as antigen-specific APCs to induce unresponsiveness or poor responses in naive T cells = (166-172)= . Although there were some controversial results demonstrating that rB cells were able to induce a T cell response, the ability of rB cells to behave as competent APCs was quite weak = (171)= . Therefore, it has been suggested that rB cells may be potent inducers of anergy both in vitro and in vivo. Data in support of this hypothesis has been observed by Eynon et al. who showed that mice injected intravenously with Fab of rabbit anti-IgD antibody, which targeted rB cells, induced tolerance to the Fab fragments after secondary challenge with the same antigen = (173-175)= . Moreover, Fuchs and Matzinger reported that pretreatment with donor male rB cells induced prolonged survival of male grafts in female recipients (H-Y mismatch) = (185)= . Therefore, rB cells may be tolerogenic under some conditions (Figure 1-2). 3.2 Results 3.2.1 FACS analysis of rB cells 3.2.1.1 Expression of B7-1 and B7-2 Resting B cells did not express B7-1 and B7-2. First, we examined the expression of MHC class II, B7-1, B7-2, surface Ig and CD40 by rB cells, aB cells, LODACs, peripheral blood mononuclear cells (PBMCs) and unfractionated splenocytes using FACS analysis (Table 3-1). In PBMC (i.e. component of DST) 58% of leukocytes were surface Ig+ and 67.7% were MHC class II+. Only 1.9% and 2.5% of cells expressed B7-1+ and B7-2+ cells, respectively (Table 3-1). When unfractionated splenocytes were analysed by flow cytometry 66% of leukocytes were MHC class II+ and 64.6% surface Ig+. These percentages were the same as in the PBMC population (67.7% and 58%, respectively). B7-1 and B7-2 expression in unfractionated splenocytes was much higher than found in PBMC (4.2% and 8.0%, and, 1.9% and 2.5%, respectively) (Table 3-1). Resting B cells purified as described in Section 2.5.2 expressed MHC class II (95.7%) and surface Ig (92.3%), but expressed low levels of B7-1 and B7-2 (1.2% and 2.5%, respectively). After culturing the rB cells with LPS for 2 days, B7-1 and B7-2 were expressed strongly (49.5% and 77.7%, respectively) (Table 3-1). Although the percentage of MHC class II+ cells in the B cell population was the same before and after LPS activation (95.7% and 96.1%, respectively), the mean channel fluorescence increased after activation (data not shown). LODACs purified as described in Section 2.5.3 expressed MHC class II, B7-1 and B7-2 strongly, (92.2%, 68.8% and 87.2%, respectively). The contamination of B cells in the LODACs population was 9.4%. 3.2.1.2 Expression of B7-1 and B7-2 after LPS activation B7-1 and B7-2 were expressed on rB cells after activation with LPS. Next, we examined the kinetics of B7 expression on rB cells after LPS activation. As demonstrated by monitoring of the percentage of positive cells, B7-1 and B7-2 expression on B cells were maximal on day 2 and day 3 after LPS activation, respectively (75% and 35%). When examined using mean channel fluorescence, B7-1 expression on B cells was maximal 2 days after activation and B7-2 expression on B cells increased during the 4 day observation period (Figure 3-1). As LPS upregulated B7 expression, all tissue culture medium was checked for LPS contamination by the Therapeutic Antibody Centre, Oxford in order to avoid unexpected upregulation of B7 on cells used in this study. 3.2.1.3 Expression of B7-1 and B7-2 after stimulation of allogeneic T cells B7-1 and B7-2 were expressed on rB cells after culture with allogeneic T cells. When rB cells were cultured with allogeneic T cells in vitro, expression of B7-1 and B7-2 increased during the 3 day observation period (Figure 3-2). When the experiments were repeated using irradiated rB cells as stimulators, even though the majority of rB cells were dying in the culture, B7-1 and B7-2 expression increased on the residual population of viable cells (Figure 3-2). 3.2.2 MLC assay (rB cells as stimulators) 3.2.2.1 After each step in the preparation for B cells Having confirmed that the rB cells to be used in this study did not express B7 molecules, we examined their functional properties. To test the hypothesis that rB cells are incompetent APC in vitro, we asked whether rB cells were able to behave as stimulators in MLC. Three critical steps are required for the preparation of rB cells (i.e. antibody complement lysis for T cell depletion, sequential Sephadex G-10 columns for dendritic cell and macrophage depletion, and discontinuous Percoll gradient purification to separate rB and aB cells (Section 2.3.2)). We examined the ability of the cells isolated at each stage to act as stimulators of allogeneic T cells in MLC (Figure 3-3). The T cell-depleted fraction stimulated a vigorous MLC response. After the first Sephadex G-10 column, the ability of the preparation to stimulate on MLC response was reduced and this was decreased further after the second Sephadex G-10 column. Finally, cells isolated from on the 60/70% interface of the Percoll gradient were unable to stimulate allogeneic T cells in this assay. In contrast, isolated cells at the 50/60% interface were able to stimulate T cell proliferation. Therefore, cells after the three step preparation method were thought to be rB cells as they could not induce the proliferation of allogeneic T cells in vitro. 3.2.2.2 MLC response of the 60/70% interface without sequential Sephadex G-10 columns Next, we tested the necessity of using sequential Sephadex G-10 columns in the preparation of rB cells. Cells were purified as before but without use of the Sephadex G-10 columns. The cells from the 60/70% interface of the Percoll gradient were found to stimulate allogeneic T cells (Figure 3-4). Therefore, the sequential Sephadex G-10 columns were essential in depleting cells capable of stimulating allogeneic T cells. 3.2.2.3 The effect of radiation on rB cells Resting B cells in this study were unable to stimulate proliferation of allogeneic T cells when irradiated with 3000 rad before culture (Figure 3-3). In order to exclude the possibility that the ability of rB cells to act as competent APCs was sensitive to radiation = (172)= , rB cells or splenocytes from C57BL/10 mice were irradiated with 1000, 2000 or 3000 rad, and their ability to act as stimulators of allogeneic T cells in MLC was examined. Although rB cells irradiated with 1000 rad induced more T cell proliferation than those irradiated with 3000 rad, proliferation induced by 1000 rad irradiated rB cells was approximately 15 times less than that of 1000 rad irradiated unfractionated splenocytes (Figure 3-5). 3.2.2.4 Comparison of the capacity of rB cells, aB cells and LODACs to act as APCs in MLC Resting B cells were unable to induce proliferation of allogeneic T cells in vitro. (mismatched for H-Y, multiple miH, single MHC and miH+MHC antigens) Having developed the purification protocol for isolating rB cells we compared the ability of different APC populations to stimulate T cells in vitro when mismatched for miH and / or MHC antigen(s). 3.2.2.4 (a) A single miH barrier, H-Y First we investigated APCs and T cells mismatched for a miH antigen. Male LODACs were able to stimulate proliferation of female T cells effectively in the C57BL/10 strain. Male splenocytes were found to be able to elicit some proliferation of female T cells. However, male rB cells were unable to induce stimulation of female T cells (Figure 3-6). 3.2.2.4 (b) Multiple miH barrier CBA and C3H mice have the same MHC class I and class II molecules (H2k haplotype) but are mismatched for multiple miH antigens. Resting B cells from C3H mice were unable to induce proliferation of T cells from CBA mice. However, LODACs from C3H mice were able to induce proliferation of CBA T cells (Figure 3-7). 3.2.2.4 (c) A single MHC barrier Next, we examined the ability of APCs to induce proliferation of T cells mismatched for a single MHC class I antigen, Kb. CBK (H2k + Kb) rB cells were unable to induce proliferation of T cells from CBA mice, although splenocytes from CBK mice induced substantial T cell proliferation (Figure 3-7). 3.2.3.4 (d) Multiple miH+MHC barrier Finally, we confirmed that rB cells were unable to stimulate proliferation of fully allogeneic T cells. LODACs were more effective in stimulating T cell proliferation than aB cells (Figure 3-9). 3.2.2.5 The effect of preculture with allogeneic rB cells T cells precultured with allogeneic rB cells were unable to respond following secondary stimulation. As rB cells were unable to induce proliferation of T cells mismatched for single miH, multiple miH, single MHC or multiple miH+MHC antigens, next we investigated the ability of rB cells to induce T cell "anergy". 1x 107 nylon wool purified C3H T cells were precultured for 4 days with either irradiated LODACs (5x 106) or rB cells (2x 107) from C57BL/10 mice. 2.5x 105 viable cells from these cultures were then incubated with irradiated LODACs from C57BL/10 mice for a further 3 days. It was demonstrated that T cells precultured with rB cells were unable to be restimulated by LODACs from C57BL/10 mice (Figure 3-10). However, preculture with LODACs resulted in a vigorous secondary response upon restimulation. This experiment was then re-examined (Figure 3-11). T cells were cultured with rB cells and rested for 2 days. These T cells failed to proliferate after subsequent stimulation of splenocytes. After the 2 day preculture of 2x 107 T cells, 4.8x 106 viable T cells were recovered after exposure to splenocytes, while only 1.2x 106 T cells remained after preculture with rB cells. Following the 2 day rest period 3x 106 and 0.5x 106 viable T cells were recovered from the splenocyte and rB cell precultures, respectively. Therefore, in both experiments, T cells precultured with allogeneic rB cells were unable to respond following secondary stimulation. Thus, rB cells might indeed produce T cell anergy. However, the possibility that rB cells induce death of alloreactive T cells could not be excluded based on these experiments. 3.2.3 CTL assay Resting B cells were unable to generate cytotoxic T cells. The cytotoxic activity of T cells after culture with rB cells, aB cells or LODACs was also examined. Resting B cells were unable to generate cytotoxic activity. LODACs and aB cells generated cytotoxic T cells (Figure 3-12). Therefore, with respect to T cell effector activity, rB cells seem to be incompetent APCs. 3.2.4. ELISA T cells cultured with allogeneic rB cells did not produce IL-2 and IFN-g. The secretion of cytokines such as IL-2, IFN-g and IL-4 in the culture supernatant of an MLC using different APCs was examined. LODACs induced secretion of both IFN-g (top panel of Figure 3-13) and IL-2 (bottom panel of Figure 3-13) at a higher level than aB cells. However, IL-2 and IFN-g were not detected in the supernatant of C57BL/10 rB cells and CBA T cells. IL-4 was not detected in any of the culture supernatants. 3.3 Discussion Resting B cells were purified using a three step protocol. Antibody-complement lysis for depletion of T cells, sequential Sephadex G-10 columns for depletion of adherent cells and Percoll gradient purification for separating rB and aB cells. Resting B cells purified using this protocol were MHC class II+ and surface Ig+ (Table 3-1). Contaminating cells were thought to be T cells which survived the antibody-complement lysis. CD3+ cells in rB cell population were usually 4-6% (Section 2.5.2). Expression of B7-1 and B7-2 by the rB cells preparation was examined by FACS analysis. The background (i.e. cell without staining) in the FACS assays performed in this study was approximately 1%. The level of B7-1 expression by the rB cells was on average 1.2%; rB cells were therefore considered negative. B7-2 expression was 2.5%. Thus rB cells may express low levels of B7-2, as has been reported in other studies = (233)= . On the other hand, aB cells strongly expressed B7-1 and B7-2 (49.5% and 77.7%, respectively) (Table 3-1). CD3+ cells contaminating the aB cell preparation were less than 2% (Section 2.5.2) (i.e. less than in the rB cells). This suggests that T cells do not survive culture with LPS. LODACs are a population of cells enriched for dendritic cells. 70% of the cells present in the LODACs preparations used in this study were N418+ (anti-CD11c, one of the better markers for dendritic cells). Therefore, dendritic cell purity was thought to be approximately 70%. Contaminating B cells were approximately 10% of this preparation (Section 2.5.3). CD40 molecules are thought to be expressed consistently on MHC class II+ leukocytes. Many studies have shown recently that CD40 is an important molecule for inducing the expression of the B7 family on APCs (Figure 1-3). In our studies, rB cells, aB cells and LODACs expressed CD40 strongly with 82.4%, 96.6% and 87.2% expression, respectively (Table 3-1). As LPS was used to prepare aB cells from the rB cell population, the kinetics of B7 expression on rB cells after LPS activation was examined. B7-1 and B7-2 were expressed on B cells with increasing frequency over 3 days (Figure 3-1). B7-2 was expressed more rapidly than B7-1 on rB cells. The kinetics of B7 expression after LPS activation was the same as other investigators have reported previously = (239, 256)= . In the MLC assay, allogeneic T cells are thought to stimulate B cell activation. Therefore, we examined the B7 expression on rB cells during culture with allogeneic T cells. The percentage expression of B7-1 and B7-2 on B cells increased gradually over the first 3 days (Figure 3-2). Resting B cells expressed B7-2 more rapidly than B7-1. Therefore, allogeneic T cells were as effective in stimulating the expression of B7 molecules on rB cells as LPS. After culturing rB cells for 2 days with either LPS or allogeneic T cells, these B cells expressed B7-1 and B7-2 at high levels. Our observations about the kinetics of B7 expression after allo-MLC are in agreement with data reported by other investigators = (233)= . Resting B cells in this study were unable to stimulate allogeneic T cell proliferation when irradiated with 3000 rad before culture. However, it could be argued that the ability of rB cells to induce proliferation was radiosensitive. Radiation might mask the possibility of rB cells to induce proliferation. Some reports suggested that rB cells irradiated with 1000 rad were able to present antigen, but, this function was greatly diminished when they were irradiated with 3300 rad = (169, 172)= . Therefore, the ability of rB cells to induce allogeneic T cell proliferation following radiation with either 1000, 2000 or 3000 rad was examined. Although rB cells irradiated with 1000 rad induced more proliferation than those which received 3000 rad, the response was approximately 15 times less than that of splenocytes irradiated with 1000 rad (Figure 3-5). This proliferation might be dependent on the survival of rB cells as 1000 rad may not stop proliferation of B cells. Therefore, rB cells are thought to be incompetent APCs. In all of the strain combinations examined, rB cell were unable to induce T cell proliferation (Figure 3-6, 7, 8, and 9). However, even irradiated rB cells expressed B7 molecules strongly 2 days after culture with allogeneic T cells (bottom panel of Figure 3-2). Despite this, the B7+ B cells could not induce the proliferation of allogeneic T cells in the last 2 days of the culture period. One possibility may be that two days of culture with B7+ B cells was not long enough to induce T cell proliferation. Another possibility is that rB cells might render T cells anergic during the B7- period and therefore the T cells could not be stimulated by B7+ B cells in the last 2 days of culture. Some experimental data support the view that incompetent APCs were not only unable to stimulate T cells, but also rendered T cells anergic (Figure 1-2) = (357)= . If T cells were rendered anergic, they would not proliferate after a second stimulation with competent APCs. In order to answer this question, two experiments were devised. Basically, T cells were precultured with either allogeneic rB cells or competent APCs, and restimulated with subsequent competent APCs. A 4 or 2 day culture period was chosen for the first culture as the majority of the T cells died over a 7 day culture (data not shown). A 3 day culture period was also chosen for second culture as T cells precultured with competent APCs showed maximal proliferation on day 3 instead of day 4 in the first MLC (data not shown). In the first experiment T cells spontaneously proliferated without second stimulation immediately after preculturing with LODACs (Figure 3-10). Therefore, in the second experiment, T cells were rest for 2 days between the two cultures. These two experiments demonstrated that T cells precultured with rB cells were unable to be stimulated with competent APCs at a later stage (Figure 3-10 and 11), a phenomenon best described as T cell anergy (Figure 1-2). However, one has to consider the possibility that the alloreactive T cells died after the first culture as only approximately 10% of cells survived the preculture period (Figure 3-11). Activation-induced cell death (AICD) may be the explanation = (206, 358, 359)= . 3.4 Summary of results rB cells, aB cells and LODACs were purified and characterized. Resting B cells were found to express MHC class II molecules but not B7-1 and B7-2. After culturing with either LPS or allogeneic T cells, rB cells expressed B7 molecules at the cell surface. These molecules are important candidates for delivering costimulatory signals to responding T cells. Resting B cells were unable to stimulate the proliferation of allogeneic T cells or generate cytotoxic T cells in vitro. Moreover, T cells precultured with allogeneic rB cells were unable to respond to subsequent stimulation by allogeneic LODACs, suggesting that allogeneic rB cells might anergize the responding T cells under these conditions. Thus, rB cells can be considered to be tolerogens in vitro. 3.5 Figures and Table (Table 3-1)
Resting B cells, LODACs, PBMC and unfractionated splenocytes were prepared from C57BL/10 mice (Section 2.5.1, 2 and 3). Resting B cells (1x 106 cells/ml) were cultured with LPS (30オg/ml) for 2 days. Cells were stained with B21.2 followed by goat anti-rat Ig FITC (anti-MHC class II), goat anti-mouse Ig FITC (whole molecule), B7-1 FITC, B7-2 FITC or CD40 FITC (Section 2.6.1). (Figure 3-1)
Resting B cells (1x 106 cells/ml) were prepared from CBA mice and cultured with LPS (30mg/ml) (Section 2.5.2). Cells were stained with B7-1 FITC or B7-2 FITC (Section 2.6.1). (Figure 3-2) Kinetics of B7-1 and B7-2 expression on rB cells during culture with allogeneic T cells
Non-irradiated (top panel) or irradiated (bottom panel) rB cells from C57BL/10 (H2b) mice were cultured with nylon wool purified CBA (H2k) T cells. Kb+ cells were labelled with biotinylated-TIB139 mAb (anti-Kb) followed by streptavidin-phycoerythrin. B7-1 and B7-2 expression on these Kb+ cells was determined by staining with FITC conjugated 1G10 (anti-B7-1) and GL1(anti-B7-2) mAbs, respectively (Section 2.6.1). (Figure 3-3)
Resting B cells were prepared from C57BL/10 spleens using mAb and complement lysis, sequential Sephadex G-10 columns, and Percoll gradient purification (Section 2.5.2). Varying numbers of irradiated cells from each stage of the purification were used as stimulators in MLC with 2.5x 105 nylon wool purified T cells from C3H mice as responders. Cells were cultured for 88 hours and 3H-thymidine was added for the last 16 hours of culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD and are representative of 3 independent experiments. (Figure 3-4)
Resting B cells were prepared using the standard protocol (Section 2.5.2) except the sequential Sephadex G-10 columns were omitted. Varying numbers of irradiated cells from either 50/60 or 60/70% interface of Percoll gradient purification were used as stimulators in MLC with 2.5x 105 nylon wool purified T cells from C3H mice as recipients. Cells were cultured for 88 hours and 3H-thymidine was added for the last 16 hours of culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD and are representative of 2 independent experiments. (Figure 3-5)
2.5x 105 rB cells or splenocytes from C57BL/10 mice were irradiated with 1000, 2000 or 3000 rad, and cultured with 2.5x 105 nylon wool purified T cells from CBA mice for 86 hours. 3H-thymidine was added for the last 16 hours of culture. Results are shown as the mean of triplicated cultures +/- SD. (Figure 3-6)
LODACs, splenocytes and rB cells were prepared from C57BL/10 male mice (Section 2.5.1, 2 and 3). These cells were irradiated with 3000 rad and cultured with 2.5x 105 nylon wool purified T cells from C57BL/10 female mice for 86 hours. 3H-thymidine was added for the last 16 hours of culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD and are representative of 2 independent experiments. (Figure 3-7)
LODACs and rB cells were prepared from C3H (H2k) mice (Section 2.5.2 and 3). These cells were irradiated with 3000 rad and cultured with 2.5x 105 nylon wool purified T cells from CBA (H2k) mice for 86 hours. 3H-thymidine was added for the last 16 hours of culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD. (Figure 3-8)
Resting B cells were prepared from CBK mice and splenocytes were prepared from CBK (H2k+Kb) and C57BL/10 (H2b) mice (Section 2.5.1 and 2). These cells were irradiated with 3000 rad and cultured with 2.5x 105 nylon wool purified T cells from CBA (H2k) mice for 86 hours. 3H-thymidine was added for the last 16 hours in culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD and are representative of 2 independent experiments. (Figure 3-9)
Resting B cells, aB cells or LODACs were prepared from C57BL/10 (H2b) mice (Section 2.5.2 and 3). These cells were irradiated with 3000 rad and cultured with 2.5x 105 nylon wool purified T cells from C3H (H2k) mice for 86 hours. 3H-thymidine was added for the last 16 hours in culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD and are representative of 20 independent experiments. (Figure 3-10)
2x 107 nylon wool purified CBA T cells (Section 2.5.4) were precultured for 2 days with 1x 108 irradiated Ficoll-purified splenocytes or rB cells from C57BL/10 mice (Section 2.5.1 and 2). Viable cells were collected using Ficoll gradient centrifugation and rested for a further 2 days. 2.5x 104 viable cells from these cultures were then restimulated with irradiated C57BL/10 splenocytes for 3 days. 3H-thymidine was added for last 16 hours of the culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD. After the 2 day preculture viable T cells were counted. 4.8x 106 T cells were recovered from the preculture with splenocytes, while only 1.2x 106 T cells remained after preculture with rB cells. Following the 2 day rest period 3x 106 and 0.5x 106 viable T cells were recovered from the splenocyte and rB cell precultures, respectively. (Figure 3-12)
1x 107 nylon wool purified CBA T cells (Section 2.5.4) were cultured for 4 days with irradiated LODACs (0.5x 107), aB cells (1x 107) or rB cells (2x 107) from C57BL/10 mice (Section 2.5.2 and 3) and tested for killing on 51Cr-labelled RMA cells as specific targets. Results are shown as the mean of triplicated cultures +/- SD. (Figure 3-13)
2.5x 105 nylon wool purified T cells from CBA mice (Section 2.5.4) were cultured with varying numbers of either irradiated rB cells, aB cells or LODACs (Section 2.5.2 and 3) for 70 hours. IFN-g (top panel) or IL-2 (bottom panel) level in the supernatant was quantified by ELISA (Section 2.9). IL-4 was not detected in any of supernatants. Chapter 4. Intravenous injection of allogeneic resting B cells 4.1 Introduction 4.1.1 Aim of this chapter In this chapter, we confirmed the results of Fuchs and Matzinger, in which pretreatment of female mice with male rB cells resulted in prolonged survival of H-Y mismatched skin grafts = (185)= . Furthermore, we extended this finding by investigating the ability of rB cells to induce unresponsiveness across more extensive histocompatibility barriers using skin and cardiac allografts as test grafts. 4.1.2 Background APCs are thought to be responsible for triggering allograft rejection. In the early 1970s, depletion of specialised APCs, probably dendritic cells, from allografts by organ culture before transplantation was shown to prolong allograft survival = (105, 176)= . In the early 1980s, several studies in different experimental systems showed that removal or inactivation of allostimulatory activity in transferred lymphoid cells enhanced their tolerogenic effect = (181, 182)= . As shown in Chapter 3 not all MHC class II+ leukocytes are potent immunostimulatory cells. Indeed rB cells have been shown by ourselves and others to be incompetent APC in vitro. At the time the work presented in this thesis began, Fuchs and Matzinger = (185)= had also reported data suggesting that rB cells might be potent tolerogens in vivo. Infusion of purified male rB cells into naive female C57BL/10 mice induced tolerance to the H-Y alloantigen as demonstrated by the indefinite survival of H-Y mismatched skin grafts. H-Y specific cytotoxic T cells could not be recovered from tolerant mice. Although pretreatment of naive recipients with rB cells was clearly shown to be tolerogenic, tolerance could not be induced in mice that were previously primed to H-Y with unfractionated splenocytes. In light of the tolerogenic potential of rB cells, we tried to extend this approach to more complicated histocompatibility barriers using skin and cardiac allografts as our test grafts. 4.2 Results 4.2.1 Skin graft prolongation following pretreatment with donor rB cells 4.2.1.1 Immunogenicity of the H-Y antigen H-Y is immunogenic in C57BL/10 but not CBA and CBK mice. First, we examined the immunogenicity of the H-Y antigen in different strains. Untreated CBA (H2k), CBK (H2k + Kb) or C57BL/10 (H2b) female mice were transplanted with male skin of the same strain = (360)= . Female CBA and CBK mice were unable to reject male skin grafts (median survival time (MST) >100 days) = (352)= . The size and appearance of skin grafts in these mice were indistinguishable from female skin graft controls. In contrast, four out of five female C57BL/10 mice rejected male skin grafts (MST = 31 days) (Figure 4-1). Although one female C57BL/10 recipient accepted male skin for over 100 days, the size of the skin graft was significantly reduced. 4.2.1.2 Induction of unresponsiveness to H-Y (C57BL/10 male to female) Resting B cells induced indefinite survival of skin grafts mismatched for H-Y. Before embarking upon an extensive series of experiments we felt that it was important to confirm Fuchs and Matzinger's results = (185)= . We reproduced the condition of their experiments exactly by using the same strain of mice, dose of rB cells and interval between pretreatment and skin grafting. Female C57BL/10 (H2b) recipients were pretreated with 1x 107 male rB, 1x 107 male activated (aB) cells or 5x 105 male LODACs 7 days before grafting with male C57BL/10 skin. As Fuchs and Matzinger showed, male rB cells induced indefinite prolongation of H-Y mismatched skin grafts in all recipients (MST > 100 days) (Figure 4-2). However, the size of the skin grafts decreased during the investigation period of 100 days. Pretreatment with LODACs accelerated skin graft rejection (MST = 10.5 days), while pretreatment with DST was ineffective at inducing graft prolongation (MST = 31.5 days) compared to naive female mice (MST = 31 days) = (361)= . The grafts were considered rejected at the time of complete destruction of graft (Section 2.13.6). Thymectomized female recipients also accepted male skin when pretreated with male rB cells (MST > 100 days) (Figure 4-2). Pretreatment with male aB cells also induced indefinite graft prolongation in 60% of recipients (MST >100 days) (Figure 4-2). 4.2.1.3 Second skin grafts in H-Y mismatched recipients Second skin grafts were rejected by recipients with long-term surviving H-Y mismatched skin grafts. As male rB cells induced prolongation of H-Y mismatched skin grafts in all recipients (Figure 4-2), we examined whether operational tolerance was established in these mice. Female recipients pretreated with rB cells which accepted H-Y mismatched skin grafts for 100 days were transplanted with a second male skin graft. These second skin grafts were rejected in the recipients (MST = 20 days), while the original grafts continued to survive for another 100 days (Figure 4-3). Similar results were obtained when second skin grafts were transplanted into female recipients which had been thymectomized before the first skin grafts (Figure 4-3 and 4). Therefore, male rB cells were able to induce graft prolongation but not operational tolerance. 4.2.1.4 H-Y mismatched skin grafts in sensitized recipients Resting B cells were unable to induce the prolongation of H-Y mismatched skin grafts in sensitized recipients. As male rB cells induced prolongation of H-Y mismatched skin grafts in all recipients (Figure 4-2), we examined whether male rB cells induced the same effect in sensitized recipients. Mice were sensitized intraperitoneally with 1x107 male splenocytes two weeks before transplantation of a male skin graft. Pretreatment with male rB cells 7 days before transplantation in these sensitized recipients was ineffective at prolonging graft survival compared to untreated control (MSTs = 30 and 31 days, respectively) (Figure 4-5). 4.2.1.5 Induction of unresponsiveness to multiple miH antigens and MHC antigens (C3H to CBA) and (C57BL/10 to CBA) Resting B cells were unable to induce the prolongation of skin grafts mismatched for multiple miH antigens or MHC antigens. Next, we examined the ability of rB cells to induce prolongation of grafts mismatched for multiple miH antigens and miH+MHC antigens. CBA (H2k) mice were pretreated with either 1x 107 rB cells or 5x 105 LODACs from C3H (H2k) mice 7 days before grafting with a C3H skin. Both groups rejected their grafts (MST = 14 and 12.5 days, respectively) (Figure 4-6). Therefore, pretreatment with donor rB cells was ineffective at inducing prolongation of multiple miH mismatched skin grafts. Not surprisingly, rB cells were also incapable of inducing prolongation of grafts mismatched for multiple miH+MHC antigens. CBA (H2k) mice were pretreated with either 1x 107 rB cells or 5x 105 LODACs from C57BL/10 (H2b) mice 7 days before transplantation of a C57BL/10 skin. Both groups rejected the skin grafts (MST = 11 and 10 days, respectively) (Figure 4-7). Therefore, donor rB cells were able to induce prolongation of skin grafts mismatched only for H-Y antigen but not multiple miH or multiple miH+MHC antigens. 4.2.2 Heart graft experiments following pretreatment with donor rB cells As rB cells but not LODACs were able to induce prolongation of skin grafts mismatched for H-Y antigen (Figure 4-2) we next extended the study to cardiac grafts. 4.2.2.1 Timing of pretreatment with DST DST was most effective when delivered 14 days before cardiac transplantation For the first study we chose 7 days as the interval between pretreatment and skin grafting as this had been reported to be effective in previous experiments = (185)= . However prior to extending this study to cardiac grafts we reexamined the optimal timing of pretreatment relative to transplantation. C3H (H2k) mice were pretreated with 0.25ml DST 0, 7, 14, or 28 days before transplantation of a C57BL/10 (H2b) heart. C57BL/10 cardiac grafts were rejected by naive C3H recipients (MST = 10 days) (Figure 4-8). Pretreatment with a DST 14 days before transplantation was found to be more effective than pretreatment at 0, 7, and 28 days before transplantation (MST = 24, 15, 11 and 12 days, respectively) (Figure 4-8). Therefore pretreatment was given 2 weeks before cardiac transplantation in this series of experiments. 4.2.2.2 Dose of rB cells required to induce unresponsiveness Pretreatment with 1x 107 rB cells was most effective at inducing cardiac graft prolongation. Previous observations in this laboratory have shown that 0.25ml of whole blood was could induce cardiac graft prolongation (unpublished data). As rB cells were to be used as the source of alloantigen, we next examined the dose of donor rB cells necessary to induce graft prolongation. When rB cells from C57BL/10 mice were used as the pretreatment inoculum, 1x 107 cells were found to be most effective at prolonging cardiac graft survival compared to 1x 106, 5x 105 and 1x 105 rB cells (MST = 22, 9, 9 and 9 days, respectively) (Figure 4-9). Therefore, 1x 107 rB cells was chosen as the pretreatment dose for subsequent studies. Although 1x 107 LODACs should have been used as pretreatment ideally, we used 5x 105 LODACs as only 2x 105 LODACs were prepared from one C57BL/10 spleen. 4.2.2.3 Unresponsiveness to H-Y mismatched cardiac allografts (C57BL/10 male to female) Resting B cells and LODACs induced indefinite prolongation of heart grafts mismatched for H-Y antigen. To extend the skin graft study to cardiac grafts, female C57BL/10 (H2b) mice were pretreated with 1x 107 male rB cells, 1x 107 male unfractionated splenocytes, 5x 105 male LODACs, or 0.25ml DST 14 days before cardiac transplantation. In contrast to H-Y mismatched skin grafts which were accepted indefinitely only following pretreatment with male rB cells, H-Y mismatched cardiac allografts were accepted indefinitely by all recipients following pretreatment not only with rB cells but also LODACs and unfractionated splenocytes (MST > 200 days in both groups) (Figure 4-10). DST also induced indefinite prolongation in 80% of recipients (MST > 200 days). Naive female mice rejected male cardiac grafts (MST = 42 days). However, one naive female mouse accepted a male heart for over 200 days. Therefore, even competent APCs such as LODACs can induce prolongation of cardiac grafts mismatched for H-Y antigen. 4.2.2.4 Tolerance to H-Y Second hearts were accepted in the recipients with long-term surviving H-Y mismatched heart grafts while donor type skin was rejected. Next, we examined whether operational tolerance to H-Y had been induced. Second cardiac grafts were transplanted in the neck of the recipients with long-term surviving hearts which had been pretreated with rB cells or LODACs. Both groups were able to accept the second male cardiac grafts for over 100 days (Figure 4-11). Therefore, in terms of H-Y mismatched cardiac grafts, both rB cells and LODACs were able to induce operational tolerance. Recipients which had accepted two hearts were then grafted with donor type (C57BL/10) skin. These skin grafts were rejected at the same tempo as in naive mice (data not shown). Therefore, this operational tolerance was tissue-specific. 4.2.2.5 Induction of unresponsiveness to multiple miH antigens (C3H to CBA) Resting B cells and LODACs induced indefinite prolongation of heart grafts mismatched for multiple miH antigens in 50% of recipients. As rB cells and LODACs were able to induce operational tolerance to H-Y mismatched heart grafts (Figure 4-10 and 11) we next investigated their ability to induce prolongation of cardiac grafts mismatched for multiple miH antigens. CBA (H2k) mice were pretreated with either donor 1x 107 rB cells, 1x 107 unfractionated splenocytes, 5x 105 LODACs, or 0.25ml DST two weeks before transplantation of a C3H (H2k) heart. In this case, DST and donor splenocytes induced indefinite graft prolongation in 60% of the recipients (MST > 100 days and P < 0.05 in the both groups) while naive CBA mice rejected C3H hearts (MST = 34 days). Although LODACs as well as rB cells induced indefinite graft prolongation in 50% of the recipients, these prolongations were not statistically significant compared to naive group (P = 0.21 and 0.14, respectively) (Figure 4-12). 4.2.2.6 Induction of unresponsiveness to a single MHC class I antigen (CBK to CBA) Resting B cells were less effective than DST in a single MHC mismatched strain combination. As rB cells and LODACs induced indefinite prolongation of cardiac graft mismatched for multiple miH antigens in half of the recipients (Figure 4-12), we next extended this study to a MHC mismatched strain combination. First, we examined their effect in recipients mismatched for a single MHC class I antigen, CBK (H2k + Kb) to CBA (H2k). DST was more effective at inducing graft prolongation than rB cells (MST = 48 and 25 days, respectively) (Figure 4-13). LODACs were ineffective at prolonging graft survival (MST = 12 days), which was similar to that in untreated controls (MST = 13 days). 4.2.2.7 Induction of unresponsiveness to multiple miH+MHC antigens (C57BL/10 to C3H or CBA) Pretreatment with rB cells was no more effective than DST in a fully allogeneic combination. Finally, we tested the ability of rB cells to induce unresponsiveness in two fully allogeneic (multiple miH+MHC mismatched) strain combinations; C57BL/10 (H2b) to C3H (H2k) and to CBA (H2k). In the C57BL/10 to C3H strain combination, LODACs accelerated graft rejection compared to the untreated control group (MST = 8 and 10 days, respectively) (Figure 4-14). Resting B cells induced graft prolongation, however this effect was only equivalent to that obtained following pretreatment with DST and splenocytes (MST = 22, 24 and 20.5 days, respectively). In the C57BL/10 to CBA strain combination, naive CBA mice rejected C57BL/10 cardiac grafts (MST = 7 days). MHC class II+ cells such as rB cells, aB cells and LODACs were all ineffective at inducing graft prolongation (MST = 8, 7 and 8 days, respectively). In contrast pretreatment with DST, splenocytes and T cells induced statistically significant graft prolongation (MST = 8, 13.5 and 13 days, respectively) compared to naive mice (P<0.05, P<0.01 and P<0.01, respectively) (Figure 4-15). 4.2.3 Characterization of resting B cells after injection 4.2.3.1 FACS 4.2.3.1 (1) Detection of donor cells after i.v. injection Donor cells could be detected in spleen for up to 24 hours after injection. Monoclonal antibody specific for donor MHC class II were used to detect allogeneic splenocytes or rB cells by FACS. When either 5x 105 splenocytes or 5x 105 LODACs from C57BL/10 mice were injected intravenously into CBA recipients, these cells were undetectable in recipient spleens 24 hours after injection. Therefore, we could not examine the fate of 5x 105 LODACs after i.v. injection. In contrast, 2% of total splenocytes were of donor MHC class II type 24 hours after i.v. injection of either 1x 107 splenocytes or 1x 107 rB cells from C57BL/10 mice. However, by 48 hours after i.v. injection neither splenocytes or rB cells could be detected (data not shown). The green intracellular dye, CFSE, was used as an alternative strategy to track the injected donor cells. 1x 107 C57BL/10 splenocytes were labelled with CFSE and injected into BALB/c mice. 12 hours later 1.4% CFSE+ cells could be detected in the spleen of recipient mice (Figure 4-16). However, by 36 and 50 hours after i.v. injection less than 0.5% of splenocytes were CFSE+. In mesenteric lymph nodes of recipient mice, 12 hours later 0.7% CFSE+ cells could be detected (Figure 4-16). However, by 36 and 50 hours after injection less than 0.3% of lymphocytes were CFSE+. In contrast, when syngeneic C57BL/10 mice were used as recipients, greater than 1% of cells in recipient spleens and mesenteric lymph nodes were CFSE+ at 12, 36 and 50 hours after i.v. injection. Therefore, we used these techniques to examine the fate of donor B cells one day after i.v. injection of 1x 107 cells. 4.2.3.1 (2) Expression of B7-2 on donor cells after i.v. injection into fully allogeneic recipients (a) Donor cells gated on MHC class II molecules Allogeneic rB cells in spleen expressed B7-2 molecules 24 hours after injection (C57BL/10 to CBA). CBA mice were injected intravenously with either 1x 107 rB cells or 1x 107 aB cells from C57BL/10 mice. Recipient spleens were harvested 24 hours after i.v. injection and were stained for FACS with biotinylated B21.2 (anti-donor MHC class II) and FITC conjugated anti-B7-2 mAb followed by streptavidin-phycoerythrin as a second stage. B7-2 expression on donor MHC class II+ cells was determined by FACS. Animals injected with aB cells served as positive controls. Freshly prepared rB cells were used as negative controls. Resting B cells were shown to upregulate B7-2 expression within 24 hours after i.v. injection (Figure 4-17). B7-1 has not been examined in this study as upregulation of B7-1 was slow and could not be detected clearly within 24 hours after activation (Figure 3-1). (b) Donor cells stained with CFSE before i.v. injection Allogeneic rB cells in spleen expressed B7-2 molecules 16 hours after injection (C57BL/10 to CBA). Next, we used CFSE staining to detect injected cells. C57BL/10 rB cells were labelled with CFSE before injection into CBA mice (experimental group). 16 hours after injection recipient spleens were harvested and stained with biotinylated anti-B7-2 mAb followed by streptavidin-Cy-Chrome. CFSE-stained rB cells even in the syngeneic recipients expressed B7-2 molecules 16 hours after i.v. injection compared to freshly isolated rB cells (Figure 4-18). However, CFSE-stained rB cells injected into allogeneic recipients expressed higher levels of B7-2 16 hours after i.v. injection compared to syngeneic control. Therefore, rB cells expressed B7-2 molecules after i.v. injection in allogeneic recipients, further supporting our previous result using donor MHC class II mAb to trace the donor cells. 4.2.3.1 (3) Expression of B7-2 on B cells after injection in H-Y mismatched recipient Donor cells stained with CFSE before i.v. injection Expression of B7-2 molecules on injected cells was not detected 16 hours after i.v. injection (male to female in C57BL/10). Despite the possibility that CFSE labelling before i.v. injection might induce B7-2 expression on rB cells in vivo it was the only strategy that was suitable for detecting donor cells in a H-Y mismatched strain combination. Therefore, C57BL/10 male rB cells were labelled with CFSE before injection into either male (syngeneic control) or female (experimental group) C57BL/10 mice, while CFSE-stained C57BL/10 male aB cells were injected into male C57BL/10 recipients (positive control). The expression of B7-2 on CFSE-labelled rB cells 16 hours after i.v. injection into either female or male recipients was the same (Figure 4-19). Therefore, injection of rB cells across a H-Y barrier did not upregulate B7-2 expression on rB cells compared to syngeneic group. 4.2.3.2 Responsiveness of recipient T cells after pretreatment with rB cells MLC--------Allogeneic rB cells become capable of stimulating T cell proliferation after i.v. injection. To examine the in vivo functional ability of rB cells after i.v. injection, 1x 107 rB cells or 1x 107 unfractionated splenocytes from C57BL/10 mice were injected into naive CBA recipients. 7 days later, the responsiveness of recipient CBA splenocytes was examined in MLC. Splenocytes from recipients pretreated with C57BL/10 rB cells showed enhanced proliferation to C57BL/10 stimulators when compared to splenocytes from mice pretreated with unfractionated C57BL/10 splenocytes (Figure 4-20). Therefore, rB cells were more immunogenic in vivo than the same number of unfractionated splenocytes after i.v. injection. CTLp--------The number of CTLp in the spleens of mice pretreated with allogeneic rB cells was higher than those pretreated with unfractionated allogeneic splenocytes. To examine the functional ability of rB cells after i.v. injection, 1x 107 C57BL/10 rB cells or 1x 107 C57BL/10 unfractionated splenocytes were injected into naive CBA recipients. 7 days later, the frequency of CTLp in recipient CBA splenocytes was examined in a limiting dilution assay. In three independent experiments pretreatment with C57BL/10 rB cells produced greater numbers of H2b specific CTLp than pretreatment with unfractionated C57BL/10 splenocytes. The ratio of alloreactive precursors per million splenocytes from mice pretreated with rB cells vs. unfractionated splenocytes was approximately 3 (Figure 4-21). Therefore, pretreatment with allogeneic rB cells induced more alloreactive CTLp compared to pretreatment with allogeneic unfractionated splenocytes, suggesting that allogeneic rB cells were immunogenic in vivo. 4.3 Discussion First, we examined the immunogenicity of the H-Y antigen in several strains of mice and found that male skin grafts were rejected in C57BL/10 (H2b) mice but not CBA (H2k) or CBK (H2k + Kb) mice. Therefore, rejection of H-Y mismatched skin grafts occurred on a H2b background but not on a H2k background. Lack of rejection in CBK mice demonstrated that the presentation of H-Y derived peptides on H2-Kb was alone unable to elicit rejection (Figure 4-1). The immunogenicity of H-Y antigen was dependent on the strain of the recipient as has previously been reported by others = (352, 362, 363)= . This might be due to possibilities that H2k mice present different, possible less immunogenic, peptides of H-Y antigen or that different repertoire is selected on H2k as opposed to H2b. We were able to confirm Fuchs and Matzinger's observation that rB cells but not LODACs induced prolongation of H-Y mismatched skin grafts in C57BL/10 mice (Figure 4-2). However, we observed that the size of the skin grafts considerably decreased with time after transplantation. Moreover, second male skin grafts were rejected in the female recipients with long-term surviving primary skin grafts at a similar tempo (10 to 40 days) to male skins rejected by naive female recipients (Figure 4-3). This might be due to recent thymic emigrants which would be generated after pretreatment with rB cells and would escape from the tolerogenic effect of rB cells. To exclude this hypothesis, recipient mice were thymectomized one week before pretreatment. Although thymectomized female accepted male grafts for over 100 days (Figure 4-2), the size of grafts decreased with time and second skin grafts were also rejected (Figure 4-3). Therefore, recent thymic emigrants were not involved in the decrease of the original graft size and the rejection of the second grafts. The original skin grafts persisted for a further 100 days after the second skin grafts were transplanted despite rejection of the second grafts (Figure 4-3 and 4). It is possible that the original grafts may have become less immunogenic after grafting. Loss of immunogenicity with time after transplantation has been reported previously = (364)= and may be due to loss of APC from the grafts = (72)= . Although male rB cells induced prolongation of H-Y mismatched skin grafts in naive mice for over 100 days (Figure 4-2), this protocol were ineffective at prolonging grafts in recipients sensitised with intraperitoneal injection of male splenocytes (Figure 4-5). This in vivo observation corresponds to in vitro results of Fuchs and Matzinger = (185)= . (1) Immunogenicity of the graft; skin is the most immunogenic graft = (366-368)= .
First to investigate the immunogenicity of the graft we compared the effects of rB cells with cardiac rather than skin grafts = (372)= . Skin grafts are thought to be more immunogenic than cardiac grafts for a number of reason including tissue-specific antigens = (367, 373)= , more competent APCs such as Langerhans cells and dendritic cells, = (368)= and lack of blood supply immediately after grafting = (374)= . Donor rB cells induced indefinite graft survival in all recipients in the H-Y mismatched combination (Figure 4-10) and in 50% of recipients mismatched for multiple miH antigens (Figure 4-12). However, donor LODACs were as effective as rB cells in prolonging the survival of cardiac allografts in this situation. To explain why donor LODACs induced prolongation of cardiac but not skin grafts mismatched for miH antigen(s) as effectively as rB cells, the following hypotheses could be considered. (1) In the MHC matched but miH mismatched strain combinations, allogeneic LODACs may not be fully activated to mature dendritic cells in vivo = (161, 162). Under these circumstances, allogeneic LODACs themselves might induce cardiac graft prolongation even though LODACs are generally thought to be immunogenic = (378-381)= .
Further experiments are required to determine the mechanisms involved. Resting B cells induced operational tolerance of H-Y mismatched cardiac grafts as second hearts were also accepted (Figure 4-11). However, this unresponsiveness was tissue-specific as these recipients rejected donor type skin, suggesting that multiple levels of peripheral tolerance exist as demonstrated by others = (383)= . When the ability of rB cells to induce unresponsiveness to MHC antigens was investigated (Figure 4-13, 14 and 15) they were found to be no more effective than DST. To explain these findings we propose that: (1) Allogeneic rB cells might become activated after i.v. injection into MHC disparate recipients.
We therefore investigated whether allogeneic rB cells might become activated after i.v. injection. Two methods were used for detecting donor cells; mAbs specific for donor MHC class II and CFSE (a green intracellular dye). Both strategies have advantages and disadvantages. The advantage using mAbs/FACS for detection of donor cells was that freshly prepared donor cells did not need to be manipulated further prior to injection, whereas CFSE/FACS permitted the detection of donor cells in the situation of a miH mismatched and MHC matched strain combination. One potential problem with this approach was that the labelling freshly prepared donor cells with CFSE and trauma of i.v. injection might induce B7-2 expression on rB cells (Figure 4-18). To try and avoid misinterpreting the data positive and negative controls for each experiment were selected very carefully. Using either method, 1 to 2% of fully allogeneic donor cells could be detected by FACS in the spleens of recipients 24 hours after i.v. injection of 1x 107 donor cells (Figure 4-16). Interestingly, B7-2 expression on allogeneic B cells was detected in vivo (Figure 4-17 and 18), suggesting that the rB cells were activated after i.v. injection into fully allogeneic recipients. In contrast, B7-2 was not expressed by rB cells after i.v. injection of male cells into female recipients compared to syngeneic control (Figure 4-19). These data were taken to indicate that H-Y antigen alone could not activate the rB cells, further supporting the hypothesis that the maintenance of the resting status of the B cells is critical for their tolerogenic capacity in vivo (Figure 4-2). These ideas were further confirmed by analysing the responsiveness of splenocytes from CBA mice pretreated with fully allogeneic C57BL/10 rB cells. These cells showed both enhanced proliferative responses and an increased frequency of CTL precursors specific for donor antigens than splenocytes from control CBA mice pretreated with fully allogeneic C57BL/10 unfractionated splenocytes (Figure 4-20 and 21). Still yet another possibility explaining the ineffectiveness of rB cell at inducing fully allogeneic graft prolongation is that rB cells might be able to anergize naive T cells but not memory T cells. This hypothesis is supported by our results that male rB cells were effective at inducing prolongation of H-Y mismatched skin grafts in naive but not sensitized recipients (Figure 4-5). T cells reactive to fully allogeneic antigen (i.e. multiple miH + MHC antigens) are thought to represent 5 to 20% of the normal T cell repertoire = (370)= . It is possible that some of these T cells might have been activated by environmental antigens (memory phenotype) = (384)= and could not be anergized by rB cells, leaving them available to respond to cross-reactive antigens on donor tissues. Antigen experienced T cells express gp39 = (268)= . Therefore, any interaction between these cells and the injected rB cells might be responsible for activation of the B cells in vivo = (114, 270, 271)= . This idea is explored in depth in the following chapter. 4.4 Summary of results Pretreatment with rB cells induced indefinite prolongation of skin grafts mismatched for single miH, H-Y, but not for multiple miH or multiple miH+MHC antigens. However, operational tolerance was not induced. In contrast, pretreatment with rB cells induced indefinite prolongation of cardiac grafts mismatched for single miH, H-Y, and multiple miH antigens but not single MHC or multiple miH+MHC antigens. Operational tolerance was demonstrated but it was tissue-specific. Interestingly, LODACs were also effective at inducing operational tolerance of cardiac grafts in miH mismatched combination. 4.5 Figures (Figure 4-1)
Naive female recipients, C57BL/10 (H2b), CBA (H2k) or CBK (H2k + Kb) were grafted with male skin from the same strain. (*P<0.05 compared with C57BL/10 group) (Figure 4-2) Protocol-1
Female C57BL/10 mice were either untreated or pretreated with 1x 107 rB cells or 5x 105 LODACs from C57BL/10 male mice one week before transplantation of a C57BL/10 male skin. Resting B cells were able to prolong the survival of skin graft indefinitely, although the size of grafted skin decreased during the period of observation. Some mice were thymectomized 2 weeks before transplantation. (*P<0.05, **P<0.01 compared with no treatment group) (Figure 4-3) Protocol-1
Naive or thymectomized C57BL/10 female recipients were pretreated with 1x 107 male resting B cells one week before transplantation of a male skin and accepted the grafts for over 100 days. Then, these female recipients were grafted again with male skin. These second skin grafts were rejected at the same tempo as a naive animals (Figure 4-2). The first grafts survived although the size of these first grafts decreased during the observation period. (**P<0.01 compared with 2nd graft groups) (Figure 4-4) Picture (Figure 4-5)
C57BL/10 female mice were sensitized intraperitoneally with 1x 107 male splenocytes two weeks before transplantation of a male skin. Naive or sensitized C57BL/10 female recipients were pretreated with 1x 107 male resting B cells one week before transplantation of a male skin. (**P<0.01 compared with sensitized group) (Figure 4-6) Protocol-1
CBA mice were pretreated with 1x 107 rB cells or 5x 105 LODACs from C3H mice one week before transplantation of a C3H skin. (Figure 4-7) Protocol-1
C3H mice were pretreated with 1x 107 rB cells or 5x 105 LODACs from C57BL/10 mice one week before transplantation of a C57BL/10 skin. (Figure 4-8)
C3H mice were pretreated with 0.25ml of DST either on the day of transplantation, or 1, 2 or 4 weeks before grafting a C57BL/10 heart. (**P<0.01 compared with no treatment group) (Figure 4-9)
Protocol-2
CBA mice were either untreated or pretreated with 1x 107, 1x 106, 5x 105 or 1x 105 rB cells from C57BL/10 mice two weeks before transplantation of a C57BL/10 heart. (*P<0.05 compared with no treatment group) (Figure 4-10)
Protocol-2
C57BL/10 female mice were either untreated or pretreated with 1x 107 rB cells, 5x 105 LODACs, 0.25ml DST or 1x 107 unfractionated splenocytes from C57BL/10 male mice 2 weeks before transplantation of a C57BL/10 male heart. (*P<0.05, **P<0.01 compared with no treatment group) (Figure 4-11) Protocol-2
C57BL/10 female mice were pretreated with either 1x 107 rB cells or 5x 105 LODACs from C57BL/10 male mice 2 weeks before transplantation of a C57BL/10 male heart. All these mice accepted their grafts for over 200 days. These mice were then challenged with donor type cardiac grafts in the neck. (Figure 4-12) Protocol-2
CBA (H2k) mice were either untreated or pretreated with 1x 107 rB cells, 5x 105 LODACs, 0.25ml DST or 1x 107 unfractionated splenocytes from C3H (H2k) mice 2 weeks before transplantation of a C3H heart. (*P<0.05 compared with no treatment group) (Figure 4-13) Protocol-2
CBA (H2k) mice were either untreated or pretreated with 1x 107 rB cells or 0.25ml DST from CBK (H2k+Kb) mice 2 weeks before transplantation of a CBK heart. (**P<0.01 compared with no treatment group) (Figure 4-14) Protocol-2
C3H (H2k) mice were either untreated or pretreated with 1x 107 rB cells, 5x 105 LODACs, 1x 107 unfractionated splenocytes or 0.25ml DST from C57BL/10 (H2b) mice 2 weeks before transplantation of a C57BL/10 heart. (*P<0.05, **P<0.01, ***P<0.001 compared with no treatment group) (Figure 4-15) Protocol-2
CBA (H2k) mice were either untreated or pretreated with 1x 107 rB cells, 1x 107 aB cells, 1x 107 T cells, 1x 107 unfractionated splenocytes, 5x 105 LODACs or 0.25ml DST from C57BL/10 (H2b) mice 2 weeks before transplantation of a C57BL/10 heart. (*P<0.05, **P<0.01 compared with no treatment group) (Figure 4-16)
1x 107 C57BL/10 splenocytes were stained with CFSE before injection and injected intravenously into either C57BL/10 or BALB/c mice. Spleens and mesenteric lymph nodes were sampled and fluorescence positive cells were counted by FACS. Data were presented as the mean of two mouse samples +/- SD and are representative of 4 independent experiments. (Figure 4-17)
Single cell suspensions from the spleens of CBA mice pretreated with 1x 107 C57BL/10 rB or aB cells were prepared 24 hours after i.v. injection. IAb+ cells were identified by labelling with biotinylated anti-IAb mAb and streptavidin-phycoerythrin. B7-2 expression on these cells was determined by staining with FITC conjugated anti-B7-2 mAb. B7-2 expression of these cells was compared to that of rB cells immediately after isolation (Section 2.6.2). (Figure 4-18)
1x 107 C57BL/10 rB or aB cells were stained with CFSE (intracellular green dye) before injecting intravenously into either CBA or C57BL/10 mice (Section 2.6.3). 16 hours later, spleens were harvested and splenocytes were stained with biotinylated anti-B7-2 mAb followed by Cy-chrome. The expression of B7-2 were examined on CFSE+ cells. (Figure 4-19)
1x 107 rB or aB cells from male C57BL/10 mice were stained with CFSE before i.v. injection into either male or female C57BL/10 mice (Section 2.6.3). 16 hours later, spleens were harvested and then splenocytes were stained with biotinylated anti-B7-2 mAb followed by Cy-chrome. The expression of B7-2 was examined on CFSE+ cells. (Figure 4-20)
1x 107 C57BL/10 rB cells or unfractionated splenocytes were injected into CBA mice. One week later, spleens were harvested and 2.5x 105 splenocytes were cultured with irradiated C57BL/10 splenocytes for 2 days and 3H-thymidine was added for the last 16 hours of culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD are representative of 3 independent experiments (Figure 4-21)
Either 1x 107 rB cells or splenocytes from C57BL/10 mice were injected into CBA mice. Two weeks later, spleens were harvested and used in a limiting dilution assay for CTLp as described in Section 2.8.4. Results are shown as the mean ratio compared to splenocytes +/- SD. Chapter 5. Blockade of gp39 / CD40 pathway 5.1 Introduction 5.1.1 Aim of this chapter Pretreatment with rB cells induced indefinite prolongation of grafts mismatched for miH but not of grafts mismatched for MHC antigens. This might be due to the activation of rB cells after i.v. injection. Recently, it has been suggested that the gp39/CD40 pathway is important for the activation of B cells. Studies have demonstrated that rB cells were tolerogens in the absence of signals through CD40 = (336)= . Therefore, we tried to use anti-gp39 mAb in combination with donor rB cells to induce prolongation of fully allogeneic cardiac grafts. 5.1.2 Background B7-1 (CD80), B7-2 (CD86) and CD40 are putative costimulatory molecules expressed on APCs = (232, 253, 351, 385-387)= . Binding of B7-1 and B7-2 to their counter-receptor, CD28, on resting T cells increases lymphokine production, promotes T cell expansion, and thereby prevents the induction of T cell anergy = (112, 357, 385, 388)= . The capacity of APCs to induce T cell activation may depend on the relative level of expression as well as the density of these costimulatory molecules. Some professional APCs have been demonstrated to express high densities of B7-1 and B7-2 molecules = (235, 237, 356)= . In contrast, rB cells possess antigen-specific Ig receptors and MHC class II molecules but do not express B7-1 or B7-2 = (191, 389)= (Table 3-1). It has therefore been proposed that rB cells are incompetent or non-professional APCs = (170, 390)= . The gp39/CD40 pathway plays an important role in B cell activation (Figure 1-3), including proliferation and immunoglobulin isotype switching = (114, 278, 391)= . CD40, a phosphorylated glycoprotein, is a member of the TNF-receptor superfamily and is expressed on B cells, endothelial cells, macrophages, dendritic cells, T cells and fibroblasts = (264, 266, 268)= . The ligand for CD40, gp39, has been identified as a 39-kDa glycoprotein expressed on activated but not resting T cells and mast cells = (268)= . gp39 mRNA transcripts have also been detected by PCR in TCR g+カ+ T cell clone, NK cells, monocytes, fetal thymocytes and small intestine = (392)= . Recently, Buhlmann et al. showed that in vivo administration of a mAb specific for gp39, MR1, combined with allogeneic B cells (mismatched for MHC and miH antigens) diminished the response to alloantigen in vitro = (336)= . Parker et al. have also shown that pretreatment of donor small lymphocytes in combination with MR1 prolonged the survival of fully allogeneic pancreatic islets = (340)= . The aim of this chapter was to investigate the role of the gp39/CD40 and B7/CD28 costimulatory pathways in the induction of hyporesponsiveness to alloantigens in vivo. 5.2 Results 5.2.1 Pretreatment with donor cells in combination with anti-gp39 (MR1) and/or CTLA4-Ig 5.2.1.1 Pretreatment with rB cells and MR1 MR1 augmented the ability of rB cells to induce long-term survival of fully allogeneic cardiac grafts. In order to try and prevent activation of the rB cells after i.v. injection, cardiac allograft recipients, C3H (H2k) mice, were pretreated with 1x 107 donor (C57BL/6) (H2b) or third party (BALB/c) (H2d) rB cells and 250オg anti-gp39 mAb, MR1, 14 days before grafting. Donor rB cells combined with MR1 induced indefinite prolongation of allograft survival (>100 days) in 45% of recipients (Figure 5-1). This effect was donor specific as rB cells from BALB/c mice in combination with MR1 were ineffective at prolonging the survival of C57BL/6 cardiac allografts (MST = 18 days). Pretreatment with donor rB cells alone was ineffective (MST = 11 days), while pretreatment with MR1 alone showed slight graft prolongation (MST = 18 days). Hearts from C57BL/6 mice were rejected when transplanted into naive C3H recipients (MST = 9 days). 5.2.1.2 B7-2 expression on rB cells after MR1 treatment MR1 did not prevent expression of B7-2 on rB cells after i.v. injection To determine whether the improvement in graft survival observed when rB cells were injected in combination with MR1 was due to inhibition of B7-2 induction, the rB cells were examined by FACS analysis. Surprisingly, B7-2 expression was detectable within 24 hours of injection even when pretreatment was performed in the presence of MR1 (Figure 5-2). 5.2.1.3 Pretreatment with rB cells and MR1 + CTLA4-Ig Pretreatment with MR1 and CTLA4-Ig showed minimal effect for inducing graft prolongation compared to MR1 alone when delivered with rB cells. As MR1 did not prevent the expression of B7-2 on allogeneic rB cells in vivo (Figure 5-2) and pretreatment with rB cells in combination with MR1 did not induce indefinite cardiac graft survival in all recipients (Figure 5-1), CTLA4-Ig was added to the pretreatment protocol in order to prevent engagement of CD28. Pretreatment with CTLA4-Ig and MR1 in combination with rB cells induced indefinite graft prolongation (> 100 days) in 66% of the recipients (Figure 5-3). However, the difference in graft survival with or without the addition of CTLA4-Ig was not statistically significant (P = 0.38). Pretreatment with CTLA4-Ig alone was ineffective when compared to graft survival in untreated recipients (MST = 11 and 9 days, respectively), while pretreatment with CTLA4-Ig in combination with either MR1 or rB cells resulted in some graft prolongation (MST = 17 and 22 days, respectively). Thus the addition of CTLA4-Ig to the pretreatment regimen did not have any significant additional effect. 5.2.1.4 Pretreatment with MHC class II positive cells and MR1 Competent APCs induced hyporesponsiveness to alloantigens when delivered with MR1. As the expression of B7-2 by rB cells did not inhibit their ability to induce hyporesponsiveness to alloantigens in vivo when delivered in combination with MR1 we then examined the ability of other APCs that express B7-1 and B7-2 constitutively to induce specific unresponsiveness. CBA (H2k) mice were pretreated with either 1x 107 aB cells, 1x 107 rB cells or 5x 105 LODACs from C57BL/10 (H2b) mice either alone or in combination with 250mg MR1 14 days before transplantation of a C57BL/10 cardiac allograft. Mice pretreated with rB cells, aB cells, LODACs or MR1 alone rejected the allografts at the same rate as untreated controls (MST = 9, 7, 8, 9 and 8 days, respectively). However, when pretreatment was combined with MR1, aB cells and LODACs as well as rB cells were found to be capable of inducing indefinite prolongation of cardiac grafts (> 100 days ) in 60, 80 and 100% of the recipients, respectively (Figure 5-4). Moreover, all cardiac allografts in mice pretreated with LODACs and MR1 beat at the same rate as syngeneic hearts as determined by ECG (Section 2.13.9), whereas the allografts in mice treated with rB cells and MR1 beat more slowly or irregularly (data not shown). In fact, as judged by histological analysis pretreatment with LODACs in combination with MR1 was the most effective strategy. Hearts from these mice had only a low number of infiltrating cells, while hearts from mice pretreated with rB cells showed higher levels of infiltration when delivered with MR1 (Figure 5-5). Taking into account heart rate and histology, aB cells were less effective than LODACs but both sources of cells were more effective than rB cells when used in combination with MR1 (data not shown). 5.2.1.5 Pretreatment with MHC class II negative cells and MR1 MHC class II+ cells were not necessary to induce graft prolongation when delivered with MR1. To investigate whether expression of allogeneic MHC class II molecules by the cells used for pretreatment was necessary, recipients were treated with MHC class II negative cells. T cells were isolated from the spleens of C57BL/10 mice by elution from a nylon wool column followed by negative sorting by FACS. In this population, contamination by MHC class II+ cells was found to be less than 0.5% (Section 2.5.5). 1x 107 of these T cells were injected in combination with MR1 into CBA mice two weeks before transplantation of a C57BL/10 heart. Although all recipients treated with these T cells alone rejected their allografts (MST = 13 days), 66% of the recipients accepted their grafts indefinitely (> 100 days) when T cells were delivered with MR1 (Figure 5-6). Secondly, CBA mice were pretreated with 1x 107 splenocytes from CBK transgenic mice (H2k + Kb) in combination with MR1 = (343)= . In this case, 66% of the recipients accepted C57BL/10 grafts indefinitely (> 100 days). CBK splenocytes alone were ineffective at prolonging graft survival (MST = 7 days) (Figure 5-6). Therefore, expression of allogeneic MHC class II molecules by the antigen inoculum was not essential for the induction of unresponsiveness in this model. 5.2.1.6 Operational tolerance Recipients with long-term surviving heart grafts accepted secondary donor type heart transplants but not third party grafts. To investigate the development of operational tolerance in recipients with long-term surviving primary grafts induced by pretreatment with donor APCs in combination with MR1, recipient mice which had accepted C57BL/10 hearts for over 100 days were randomly assigned for transplantation of either donor type (C57BL/10) or third party (either BALB/c or F1(BALB/c x CBA)) hearts. All second donor type hearts were accepted for over 100 days, while third party grafts were rejected (MST = 16 days) (Figure 5-7). Therefore, pretreatment with donor cells in combination with MR1 had induced donor-specific operational unresponsiveness to alloantigens in vivo. 5.2.2 CD40 knockout mice as B cell donors Failure of rB cells from CD40 -/- mice to induce hyporesponsiveness to alloantigens in vivo To directly test the role of the gp39/CD40 pathway in the prolongation of allograft survival in this model (Figure 5-8), C3H (H2k) mice were pretreated with 1x 107 rB cells from CD40 -/- mice (H2b) and transplanted with wild type (C57BL/6 x 129)F1 (H2b) hearts. Pretreatment with CD40 -/- rB cells failed to prolong allograft survival (MST = 11 days) (Figure 5-9). C3H mice pretreated with (C57BL/6 x 129)F1 rB cells rejected (C57BL/6 x 129)F1 grafts at the same rate as untreated recipients (MST = 9 and 11 days, respectively). Thus CD40 -/- rB cells alone failed to induce unresponsiveness to alloantigen. 5.2.3 gp39 knockout mice as recipients Graft prolongation in gp39 -/- mice following pretreatment with allogeneic rB cells To investigate the role of gp39 expression by recipient cells, gp39 -/- mice were used as recipients (Figure 5-8). Naive gp39 -/- mice (H2b) rejected C3H (H2k) grafts acutely (MST = 8 days). Interestingly, pretreatment of gp39 -/- mice with 1x 107 rB cells from C3H mice resulted in some graft prolongation (MST = 26.5 days). (C57BL/6 x 129)F1 recipients as wild type controls, rejected C3H grafts acutely either when untreated or following pretreatment with 1x 107 C3H rB cells (MST = 8 and 13 days, respectively) (Figure 5-10). Therefore, although rB cells from CD40 -/- mice failed to induce hyporesponsiveness to alloantigens in vivo, graft prolongation was induced in gp39 -/- mice following pretreatment with rB cells. Taken together, these data confirm that blockade of the gp39/CD40 pathway in the recipient immune system is able to induce graft prolongation. 5.2.4 CD40 expression on activated T cells The gp39/CD40 pathway in recipient immune system could play a role in the induction of unresponsiveness in two ways: (i) interaction between recipient MHC class II+ cells and T cells including indirect presentation (Figure 5-11-c and d) and (ii) T-T interaction in the recipient (Figure 5-11-e). It is generally accepted that MHC class II+ cells express CD40 constitutively. However, expression of CD40 by T cells is less well characterized. To test whether T cells expressed CD40, C57BL/10 splenocytes were cultured with either PMA and ionomycin or allogeneic CBA splenocytes. Although naive T cells did not express CD40, T cells increased the expression of CD40 after activation with either allogeneic MLC or PMA/ionomycin as compared to isotype control (Figure 5-12). 5.2.5 In vitro analysis of mice pretreated with alloantigen in combination with MR1 5.2.5.1.(1) MLC assay Splenocytes from mice 14 days after pretreatment with MR1 and rB cells proliferated against allogeneic stimulation at the same level as untreated splenocytes. MR1 in combination with rB cells was able to induce prolongation of cardiac allograft survival (Figure 5-1). We therefore examined the responsiveness of recipient leukocytes in MLC in the induction phase. CBA mice were either untreated or pretreated with 1x 107 C57BL/10 rB cells, 250オg MR1 or both. Splenocytes from mice pretreated with rB cells showed higher proliferative response against irradiated allogeneic splenocytes compared to naive mice while splenocytes from mice pretreated with MR1 alone showed lower proliferative response compared to naive mice. Pretreatment with MR1 in combination with rB cells reduced the response to the same level as that found in naive mice (Figure 5-13). This result did not correlate with graft survival (Figure 5-1 and 4). 5.2.5.1.(2) CTLp assay Splenocytes from mice 14 days after pretreatment with MR1 and rB cells were unable to generate precursors of cytotoxic T cells. Next, the number of CTLp in CBA mice pretreated with 1x 107 rB cells in the absence or presence of 250オg MR1 was determined in limiting dilution assays. Pretreatment with rB cells alone increased the number of CTLp in the splenocytes, while pretreatment with rB cells in combination with MR1 dramatically reduced the number of CTLp compared to the number present in naive mice (Figure 5-14). Therefore, cytotoxicity against donor alloantigen was impaired two weeks after pretreatment with MR1 and rB cells. This result did correspond to the survival of cardiac allografts following pretreatment with MR1 and rB cells (Figure 5-1 and 4). 5.2.5.2 Maintenance phase (100 days after transplantation) 5.2.5.2. (1) MLC assay Splenocytes from mice with cardiac grafts surviving for 100 days following pretreatment with MR1 + alloantigen proliferate against allogeneic stimulators to the same extent as splenocytes from naive mice. Many studies have shown that the presence of the graft appears to contribute to the maintenance of tolerance. We therefore considered it possible that although we had failed to identify changes in MLR responses during the induction phase, changes might become apparent in mice bearing long-term survival grafts. Splenocytes from mice accepting grafts as a result of pretreatment with MR1 + LODACs showed as good a response against allogeneic donor stimulators as naive splenocytes (Figure 5-15). However, cardiac grafts were still accepted in these recipients (Figure 5-4). 5.2.5.2. (2) CTL assay Splenocytes from mice accepting cardiac grafts over 100 days pretreated with MR1/alloantigen generated cytotoxic T cells. Next, we examined whether splenocytes from mice with long-term surviving cardiac grafts could generate CTL effector cells. Although splenocytes from mice with long-term surviving grafts were less effective that naive splenocytes as responders, they were able to generate cytotoxic T cells against target cells which had the same H2 haplotype as the grafts (Figure 5-16). 5.2.5.2. (3) Alloantibody assay Alloantibody against donor cells was not detected in mice accepting cardiac grafts over 100 days following pretreatment with MR1/alloantigen The gp39/CD40 pathway is thought to play a pivotal role in the induction of humoral immunity, immunoglobulin class switching and germinal center formation. Therefore, we examined the formation of alloantibody in recipients with long-term surviving hearts following pretreatment with alloantigen and MR1. Alloantibodies specific for Kb were not detected in serum from mice with long-term surviving grafts (Figure 5-17). Mice which have rejected their grafts provide anti-Kb alloantibody in our study (data not shown) = (393)= . 5.3 Discussion Exposure to soluble antigens or peptides can result in the development of specific immunological unresponsiveness = (107, 394)= . In vivo, pretreatment with alloantigen can also reduce immune unresponsiveness to an allograft = (297)= but, in the majority of situations the unresponsiveness is incomplete, resulting in prolonged but not indefinite allograft survival. The aim of this chapter was to understand the role of costimulatory pathways in the induction of hyporesponsiveness to alloantigen in vivo. The gp39/CD40 pathway plays an important role in B cell activation = (270, 271)= , due in part to its ability to increase expression of B7 on the B cell (Figure 1-3) = (271, 281, 283)= . Thus we hypothesised that blockade of the gp39/CD40 pathway by MR1 would prevent up-regulation of B7 by the rB cells in vivo thereby augmenting their ability to induce unresponsiveness due to lack of costimulation at the time of antigen recognition. Pretreatment with rB cells in combination with anti-gp39 mAb (MR1) induced clear specific prolongation of fully allogeneic cardiac allografts (Figure 5-1). These results were consistent with those of others who have shown the ability of this strategy to inhibit alloimmune responses in vitro = (336)= and in a mouse pancreatic islet transplant model = (340)= . Surprisingly, examination of B7 expression by the allogeneic rB cells 24 hours after in vivo delivery in combination with MR1 revealed increased expression of B7-2 (Figure 5-2). Therefore, MR1 did not function in this model simply by preventing increased expression of B7-2 molecules. These findings were consistent with the reported finding by Larsen et al. that expression of B7-1 and B7-2 transcripts in cardiac allografts was not inhibited by blockade of the gp39/CD40 pathway = (279)= . Taken together, these data suggest that B7 expression in these transplant models was, at least in part, regulated by gp39/CD40 independent factor(s). These results suggested that the ability of anti-gp39 mAb to augment the hyporesponsiveness induced by rB cell treatment was not due to reduced delivery of costimulation as a result of inhibition of the up-regulation of B7 expression on the rB cells. This hypothesis was supported by the finding that pretreatment with rB cells in combination with CTLA4-Ig produced only minimal graft prolongation (Figure 5-3). This observation might be explained by incomplete blockade of the B7/CD28 interaction following a single dose of CTLA4-Ig in vivo. However, this was considered to be unlikely as CTLA4-Ig has high avidity for both B7-1 and B7-2 and it has a long serum half life = (326, 395)= . Alternatively the gp39/CD40 and B7/CD28 pathways may function independently in the response to alloantigens in vivo. This is supported by an observation that although either MR1 or CTLA4-Ig alone could not induce cardiac graft prolongation, treatment with both induced indefinite graft prolongation = (329)= . If this was the case, then an increased ability to induce hyporesponsiveness should be seen with combined blockade of the B7/CD28 and gp39/CD40 pathways when combined with rB cell treatment. Pretreatment with this combination was more effective at inducing indefinite allograft prolongation than when MR1 was used alone when delivered with allogeneic rB cells (Figure 5-3). However, the difference was not dramatic and not statistically significant (p=0.38) (Figure 5-3). While the reasons for the minimal effect of this combination treatment, as compared to others = (329)= are not clear, the results suggested a dominant role for the gp39/CD40 pathway in determining the response to alloantigen in this system. Taken together, the results suggest that blockade of the B7/CD28 pathway does not dramatically augment the ability of donor antigen to induce specific hyporesponsiveness. To determine if B7 expression prevented this effect, cells that constitutively express B7 were tested in this model. Pretreatment with both LODACs and aB cells in combination with MR1 resulted in indefinite prolongation in 100 and 80% of the recipients, respectively (Figure 5-4). Moreover, LODACs in combination with MR1 was found to be the most effective strategy (Figure 5-5). Thus although the B7 pathway was not irrelevant in this system, its role was independent of the gp39/CD40 pathway. Expression of B7-1 and B7-2 by cells present during alloantigen pretreatment did not abrogate the induction of unresponsiveness induced by blockade of the gp39/CD40 pathway at the time of antigen recognition. Therefore, the CD40/gp39 signal between donor APCs and recipients T cells (i.e. direct presentation) may act as costimulatory pathway independent of the B7/CD28 pathway (Figure 1-3) (Figure 11-b). On the other hand, MHC class II negative cells (i.e. T cells and CBK splenocytes) also induced indefinite graft prolongation in 66% of the recipients in this model (Figure 5-6). As administration of MHC class II positive cells gave better results than MHC class II negative cells when delivered with MR1, the possibility that the CD40/gp39 pathway can act as an independent costimulatory signal in direct presentation (Figure 11-b) can not be excluded. However, other possibilities have to be considered as the mechanism involved in our study. (i) Interaction between donor MHC class I and recipient TCR in the absence of gp39/CD40 signals may induce prolongation of fully allogeneic cardiac grafts by direct recognition (Figure 11-a). (ii) The gp39/CD40 pathway could play an important role in the interaction between recipient cells including indirect presentation of alloantigen (Figure 11-c, d, e, and f) = (71)= . To dissect these two possibilities further we next prepared rB cells from CD40 -/- mice and used gp39 -/- mice as recipients (Figure 5-8) = (396)= . The failure of pretreatment with rB cells from CD40 -/- mice to induce prolonged graft survival (Figure 5-9) suggested that blockade of the gp39/CD40 pathway between donor cells and responding T cells (Figure 11-a and b) was not important in this model. It could be argued that there may be a second ligand for gp39 on donor cells which can substitute for CD40. However, this seems unlikely as CD40 -/- mice demonstrate humoral and cell-mediated immune deficiency which is similar to that seen in gp39 -/- mice = (273)= . If a second ligand for gp39 existed, it might be expected that CD40-/- mice would have a relatively normal phenotype. Our results support the possibility (ii) that the gp39/CD40 pathway could play an important role in the interaction between recipient cells in the immune system. These interactions could occur during the process of indirect presentation of alloantigen between APCs and T cells (Figure 5-11-c) or an alloantigen independent pathway between gp39 on T cells and CD40 on recipient MHC class II+ cells (Figure 5-11-d), but could also be recipient gp39/CD40 interaction between T cells (Figure 5-11-e). CD40 is known to be expressed by T cells = (397)= . Also our results showed that T cells expressed CD40 after activation (Figure 5-12). Our observation that pretreatment with donor rB cells was more effective at inducing graft prolongation in gp39 -/- mice than in wild type, littermate controls (Figure 5-10) further supports the hypothesis that disruption of the gp39/CD40 pathway in the recipient may be an important element for the down-regulation of the immune response following alloantigen encounter in vivo (Figure 5-11-c, d, e, and f). Our results are consistent with those of others which demonstrate the importance of the gp39/CD40 pathway in T cell immune responses. The gp39/CD40 interaction is known to be an important element for the initiation of humoral immune responses to thymus-dependent antigens = (271)= , and has recently been reported to play an important role in cell-mediated immunity = (114, 278, 391)= . While the predominant effect of the gp39/CD40 pathway in some experimental systems seems to be via its effect on the up-regulation of the B7 molecules on APCs (Figure 1-3) = (391, 398)= , our results suggest that disruption of the gp39/CD40 signals between the recipient T cells may be functionally important for the down-regulation of the cell-mediated immune response to alloantigen in a B7 independent manner (Figure 5-11-e). gp39/CD40 interaction between T cells in the recipient might be autocrine in nature, where both gp39 and CD40 are expressed on the same T cell (Figure 5-11-f). This was further supported by a limiting dilution assay demonstrating that splenocytes from mice pretreated with MR1 in combination with alloantigen dramatically decreased the number of CTL precursors 14 days after pretreatment (Figure 5-14). However, the MLC response of these splenocytes was the same as untreated splenocytes (Figure 5-13). Pretreatment with MR1/alloantigens induced indefinite prolongation of cardiac grafts. Moreover, these recipients accepted indefinitely donor type but not third party hearts 100 days after transplant of the first graft (Figure 5-7), suggesting that this was operational tolerance. However, splenocytes from these mice 100 days after transplantation were able to generate cytotoxic T cells (Figure 5-16) although donor specific CTL activity was reduced by pretreatment in the induction phase (Figure 5-12). This dichotomy between in vivo graft survival and the existence of in vitro alloreactivity has been termed "split tolerance" and has been demonstrated in several experimental systems = (399, 400)= . At least two mechanisms have been proposed to account for these observations. First, it has been postulated that only high-affinity cells are tolerized in these systems, and the reactivity detected in vitro is that of low-affinity cells not relevant to graft rejection in vivo = (399)= . Alternatively, it has been proposed that the survival of allografts in tolerant animals reflects a switch in alloreactive T-cell population from Th1 predominance to Th2 predominance = (401, 402)= . What is clear is that in this system and many others, in vitro responses such as MLR and CTL are unable to provide a reflection of responses in vivo and as such are of only very limited value. In view of the fact that the gp39/CD40 pathway is thought to be important for humoral immunity = (271)= , we examined the alloantibodies in mice which had accepted cardiac grafts for over 100 days. Alloantibodies specific for donor MHC class I, Kb, were not detected in the serum of these mice (Figure 5-17). Therefore, suppression of humoral immunity might be involved in graft prolongation in this model. This result may suggest that blockade of gp39/CD40 pathway between recipient T cells and B cells might be involved in our model (Figure 11-d). In conclusion, hyporesponsiveness to alloantigens in vivo may be obtained by pretreatment with donor cells combined with blockade of the gp39/CD40 pathway by anti-gp39 mAb. These observations demonstrate the importance of the gp39/CD40 interaction, independent of the B7/CD28 pathway, in the recipient's immune response to alloantigen. Moreover, they suggest an important role for the gp39/CD40 pathway in cells of the recipient's immune system in regulating the outcome of the immune response to alloantigen in vivo (Figure 5-11-c, d, e, and f). 5.4 Summary of results Resting B cells are incompetent APCs in vitro and can induce specific unresponsiveness to single miH antigens in vivo. In contrast, rB cells are unable to induce hyporesponsiveness to MHC molecules in vivo unless the gp39/CD40 pathway is blocked. Surprisingly, anti-gp39 mAb did not prevent the activation of rB cells in vivo. Moreover, pretreatment with professional APCs did not prevent the induction of hyporesponsiveness to alloantigens in the presence of anti-gp39 mAb. Resting B cells from CD40 -/- mice were unable to induce unresponsiveness, while graft prolongation was achieved in gp39 -/- recipients pretreated with rB cells. These data suggest that gp39/CD40 interactions in the recipient play a critical role in the induction of hyporesponsiveness to alloantigens in vivo and this pathway is independent of B7-CD28 costimulation. 5.5 Figures (Figure 5-1) Pretreatment with donor rB cells induced indefinite survival of fully allogeneic cardiac grafts in 45% of the recipients when delivered with anti-gp39 mAb (MR1). Protocol-2 C3H mice were pretreated with 1x 107 rB cells from C57BL/6 mice, 250オg of anti-gp39 mAb, MR1, or both two weeks before transplantation of a C57BL/6 heart. For third party controls, BALB/c mice were used as pretreatment donor instead of C57BL/6 mice. (**P<0.01 compared with no treatment group). (Figure 5-2) Anti-gp39 mAb was unable to prevent the expression of B7-2 on rB cells after i.v. injection. Single cell suspensions from the spleens of CBA mice pretreated with 1x 107 C57BL/10 aB cells or rB cells in the presence of anti-gp39 mAb, MR1 were prepared 24 hours after i.v. injection. IAb+ cells were labelled with biotinylated B21.2 (anti-IAb) mAb followed by streptavidin-phycoerythrin. B7-2 expression on these cells was determined by staining with FITC-conjugated GL1 (anti-B7-2) mAb (Section 2.6.2). B7-2 expression on these cells was compared to that of rB cells immediately after isolation. (Figure 5-3) Combination of anti-gp39 mAb and CTLA4-Ig showed minimal effect for graft prolongation compared to anti-gp39 mAb alone when delivered with donor rB cells. Protocol-2 C3H mice were pretreated with 1x 107 rB cells from C57BL/6 mice, 250オg of anti-gp39 mAb, MR1, and/or CTLA4-Ig two weeks before transplantation of a C57BL/6 heart. (**P<0.01 compared with no treatment group) (Figure 5-4) Donor B7-2+ cells were effective at inducing indefinite graft prolongation when delivered with anti-gp39 mAb. Protocol-2 CBA mice were pretreated with 5x 105 LODACs, 1x 107 rB or aB cells from C57BL/10 mice in the presence or absence of 250オg anti-gp39 mAb, MR1, two weeks before transplantation of a C57BL/10 heart. (**P<0.01 compared with no treatment group) (Figure 5-5) (Figure 5-6) Donor MHC class II negative cells were able to induce indefinite graft prolongation when delivered with anti-gp39 mAb. Protocol-2 CBA mice were pretreated with 1x 107 T cells from C57BL/10 mice or 1x 107 splenocytes from CBK mice in the absence or presence of 250オg anti-gp39 mAb, MR1. Two weeks later the mice were transplanted with C57BL/10 hearts. (*P<0.05, **P<0.01 compared with no treatment group). (Figure 5-7) Pretreatment with alloantigen and anti-gp39 mAb were able to induce operational tolerance. Protocol-2 CBA recipients accepting C57BL/10 cardiac grafts for over 100 days following pretreatment with anti-gp39 mAb in combination with alloantigens were grafted with second cardiac grafts from either donor (C57BL/10) or third party (BALB/c or F1(BALB/c x CBA mice)). (**P<0.01 compared with the third party group). (Figure 5-8) Experiments using CD40 -/- and gp39 -/- mice Experiments using CD40 -/- mice as B cell donor and gp39 -/- mice as recipients If blockade of gp39/CD40 pathway between donor APC and recipient T cells was involved in mechanism of graft prolongation following pretreatment with alloantigen in combination with MR1, both pretreatment with rB cells from CD40 -/- mice (a) and pretreatment with rB cells in gp39 -/- mice (b) would be effective at inducing unresponsiveness to alloantigen. If blockade of gp39/CD40 pathway between recipient cells was responsible for the graft prolongation, only pretreatment with rB cells in gp39 -/- mice (a) would induce graft prolongation. (Figure 5-9) Pretreatment with rB cells from CD40 -/- mice did not induce prolonged survival of fully allogeneic cardiac grafts. Protocol-2 C3H mice were pretreated with 1x 107 rB cells from either CD40 -/- or F1(129 x C57BL/6) mice two weeks before transplantation of a F1 (129 x C57BL/6) hearts. (Figure 5-10) Pretreatment with rB cells prolonged survival of cardiac allografts in gp39 -/- but not gp39 +/+ recipients. Protocol-2 gp39 -/- or F1(129 x C57BL/6) mice were either untreated or pretreated with 1x 107 rB cells from C3H mice two weeks before transplantation of a C3H heart. (*P<0.05 compared with either naive gp39 -/- mice or wild type mice pretreated with C3H rB cells). (Figure 5-11) gp39/CD40 pathways in the immune system (a) gp39/CD40 pathway between donor MHC class I and recipient TCR (Figure 5-12) CD40 was expressed on activated T cells. Splenocytes (1x 106 cells/ml) from C57BL/10 mice were cultured with either PMA (50ng/ml) and Ionomycin (500ng/ml), or irradiated allogeneic CBA splenocytes (1x 106 cells/ml). Cells were stained with biotinylated-KT3 (anti-CD3) mAb followed by streptavidin-phycoerythrin, and either FITC-conjugated CD40 mAb or FITC conjugated-isotype control (Section 2.6.1). The CD40 expression (bold line) on CD3+ cells was compared to the isotype control (thin line). (Figure 5-13) Splenocytes from mice pretreated with allogeneic rB and anti-gp39 mAb proliferated as much as untreated splenocytes when stimulated with allogeneic splenocytes. CBA mice were either untreated or pretreated with 1x 107 C57BL/10 rB cells alone, 250オg anti-gp39 mAb (MR1) alone, or both. Two weeks later, splenocytes from these mice were cultured with varying numbers of irradiated C57BL/10 splenocytes for 62 hours. 3H-thymidine was added for the last 16 hours of culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD and are representative of 2 independent experiments. (Figure 5-14) Pretreatment with allogeneic rB cells in combination with anti-gp39 mAb reduced the number of CTL precursors in the recipient spleen. CBA mice were either untreated or pretreated with 1x 107 C57BL/10 rB cells in the presence or absence of 250オg MR1. Two weeks later, splenocytes from these mice were examined by limiting dilution assay as described in Section 2.8.4. (Figure 5-15) Splenocytes from mice with long-term functioning cardiac graft induced by pretreatment with donor LODACs and anti-gp39 mAb had essentially normal proliferative responses to donor type LODACs. CBA mice were pretreated with 5x 105 LODACs from C57BL/10 mice in combination with anti-gp39 mAb, MR1, two weeks before transplantation of a C57BL/10 heart. These mice accepted cardiac grafts for over 100 days. Splenocytes from these mice were cultured with varying numbers of irradiated C57BL/10 LODACs for 62 hours and 3H-thymidine was added for the last 16 hours of culture (Section 2.8.1). Results are shown as the mean of triplicated cultures +/- SD. (Figure 5-16) Donor specific CTL could be generated in splenocytes from mice with long-term surviving cardiac grafts. CBA mice were pretreated with 1x 107 rB cells from C57BL/10 mice in combination with anti-gp39 mAb, MR1 two weeks before transplantation of a C57BL/10 heart. These mice accepted cardiac grafts for over 100 days. Splenocytes from these mice were cultured with irradiated C57BL/10 splenocytes for 4 days and tested for killing on 51Cr labelled RMA and P815 as specific and non-specific targets, respectively (Section 2.8.3). Results are shown as the mean of triplicated cultures +/- SD. (Figure 5-17) Alloantibody was not detected in recipients with long-term surviving cardiac graft by pretreatment with donor aB cells and anti-gp39 mAb. CBA mice were pretreated with 1x 107 aB cells from C57BL/10 mice in combination with anti-gp39 mAb, MR1 two weeks before transplantation of a C57BL/10 heart. These mice accepted cardiac grafts for over 100 days. Serum from these mice was collected and examined for alloantibody (Section 2.7). 4x 105 L-Kb cells were incubated with either serum from (1) the recipients or (2) naive mice, or (3) tissue culture supernatant of TIB139 (anti-Kb mAb) for 45 minutes on ice. After washing they were incubated for an additional 45 minutes with FITC-labelled goat anti-mouse IgG. The result shown is representative of 5 animals. Chapter 6: Conclusion Discussion, implications of results and a potential strategy to induce donor specific unresponsiveness in transplantation 6.1 Introduction Donor-specific tolerance remains the holy grail of organ transplantation. Although current non-specific immunosuppressive strategies result in improved patient and graft survival in the short term = (8)= , the need for chronic life-long immunosuppression is highly significant in terms of mortality and morbidity. Donor-specific tolerance could eliminate these side effects. However, in order to overcome the shortage of organs, the use of xenografts may be a way to solve this problem and the induction of tolerance in xenotransplantation may be essential if this approach is to be accepted in the future = (61)= . With so much effort world-wide being devoted to achieve true donor-specific tolerance, it is hoped that a safe and reliable method will be developed that is effective in the clinical setting. Such a method should ideally be relatively free from side effects, could be given to patients as a short course of treatment before or at the time of transplantation (as opposed to the current life-long treatment) and could be repeated if the patient requires a re-graft or transplantation of another organ. Most importantly, such treatment must not leave the patient globally immunosuppressed. Many different strategies are being investigated and one, several, or a combination of strategies may be able to achieve this goal in the not too distant future. The aims of this thesis were to investigate (i) the potential of tolerance induction using donor rB cells, (ii) blockade of gp39/CD40 pathway in combination with alloantigen as a strategy for tolerance induction and (iii) to understand some of the mechanisms involved. It is hoped that as a result of the data presented and those of others = (279, 329-331, 340-342)= blockade gp39/CD40 pathway will be one of the strategies that can achieve the goal of donor-specific operational tolerance in humans in the future. 6.2 Brief summary of results 6.2.1 Brief summary for Chapter 3 Characterization of rB cells, aB cells and LODACs demonstrated that rB cells express MHC class II molecules but not B7-1 and B7-2. After culturing with either LPS or allogeneic T cells, rB cells expressed B7 molecules at cell surface. These molecules are important candidates for delivering costimulatory signals. Resting B cells were unable to stimulate the proliferation of allogeneic T cells or the generation of cytotoxic T cells in vitro. Moreover, T cells precultured with allogeneic rB cells were unable to respond to allogeneic LODACs, suggesting that allogeneic rB cells might anergize the responding T cells. Thus, rB cells were considered to be tolerogens in vitro. 6.2.2 Brief summary for Chapter 4 Donor rB cells were injected into recipients for skin or cardiac grafts as a pretreatment. Pretreatment with rB cells induced indefinite prolongation of skin grafts mismatched only for a single miH, H-Y, but not for multiple miH or multiple miH+MHC antigens. However, operational tolerance did not developed in this system as second skin grafts were promptly rejected. In contrast, pretreatment with rB cells induced indefinite prolongation of graft mismatched for either single miH, H-Y, or multiple miH antigens but not for single MHC or multiple miH+MHC antigens. This was shown to be operational tolerance as second cardiac grafts were accepted. One potential reason for the ineffectiveness of rB cells to prolong the graft survival might be the activation of rB cells after i.v. injection. We demonstrated that allogeneic rB cells expressed B7-2 after i.v. injection. 6.2.3 Brief summary for Chapter 5 In order to prevent the activation of rB cells in vivo, pretreatment was given in combination with anti-gp39 mAb, MR1. Hyporesponsiveness to MHC molecules in vivo was induced when the gp39/CD40 pathway was blocked as judged by long-term survival of cardiac allografts. Surprisingly, anti-gp39 mAb did not prevent the activation of rB cells in vivo. Addition of CTLA4-Ig to the pretreatment protocol did not improve graft survival. Moreover, pretreatment with professional APCs did not inhibit the induction of hyporesponsiveness to alloantigens in the presence of anti-gp39 mAb. To examine the role of gp39/CD40 pathway in this system CD40 -/- and gp39 -/- mice were used. Resting B cells from CD40 -/- mice were unable to induce unresponsiveness, while modest graft prolongation was achieved in gp39 -/- recipients pretreated with rB cells. These data suggested that gp39/CD40 interactions in the recipient play a critical role in the induction of hyporesponsiveness to alloantigens in vivo and that this pathway was independent of B7/CD28 costimulation. The effects of MR1 and the data obtained from the gp39 -/- mice suggest two possibilities of interaction between gp39 and CD40. (i) gp39/CD40 interaction between recipient APC and recipient T cell including indirect presentation. (ii) gp39/CD40 interaction between recipient T cells including autocrine interaction on T cell (i.e. gp39 and CD40 interaction on the same T cell). 6.3 Potential strategy for the induction of tolerance in transplantation Resting B cells were thought to be the most important candidates as incompetent APCs. In fact, rB cells did not express B7 molecules and were unable to stimulate allogeneic T cell proliferation and generate cytotoxic T cells in vitro. However, when rB cells were used as pretreatment to induce unresponsiveness to alloantigen in vivo, they were activated after injection in MHC mismatched recipients but not miH mismatched recipients. Therefore, the strategy of using allogeneic rB cell to induce graft prolongation was limited to miH mismatched combinations = (185)= and thus would not appear to be useful in the clinical situation. One approach would be to try and maintain rB cells as stable incompetent APCs even after injection Anti-gp39 mAb, but not CTLA4-Ig, augmented the capacity of rB cells to induce indefinite prolongation of fully allogeneic cardiac grafts. However, even under these conditions rB cells expressed B7-2 under anti-gp39 mAb treatment. Moreover we found that competent APCs also induced indefinite prolongation when delivered with anti-gp39 mAb. Therefore, rB cells no longer had any special effect in a fully allogeneic strain combination. However, the data obtained provided an insight into the mechanism of unresponsiveness to alloantigen by blockade of gp39/CD40 pathway. Donor-specific tolerance at transplantation was induced by pretreatment with a single dose of alloantigen + anti-gp39 mAb. Moreover, operational tolerance was induced as a second heart but not third party grafts were accepted 100 days after transplantation. Unresponsiveness to alloantigens was achieved when the gp39/CD40 pathway in the recipient was blocked, as gp39 -/- mice induced prolonged survival fully allogeneic cardiac grafts when pretreated with rB cells. Although the gp39/CD40 pathway was first thought to be important in humoral immunity = (271)= , this pathway has recently also been shown to be involved in T cell responses = (114, 269, 270, 391)= . Our results have contributed to this conclusion. Taken together our data suggest that blockade of the gp39/CD40 pathway may be one of the strategies which might lead to donor-specific operational tolerance following alloantigen pretreatment in humans in the future. References 1. Ott RA, Mills T, Allen B. Successful treatment of acute allograft failure using pneumatic biventricular assistance. J. Heart Lung Transplant 1991; 10: 264-268. 2. Calne RY, Thiru S, McMaster P. Cyclosporine A in patients receiving renal allografts from cadaver donors. Lancet 1978; 2: 1323-7. 3. Feduska NJ, Turcotte JG, Gikas PW. Reversal of allograft rejection with intravenous methylprednisolone "pulse" therapy. J. Surg. Res. 1972; 12: 208-215. 4. Walker RG, d'Apice AJF. Azathioprine and steroids. In: Morris PJ, ed. Kidney Transplantation: Principle and Practice. 4th ed. Saunders, W. B., Philadelphia, 1994: 202-214. 5. Starzl TE, Marchioro TL, Porter KE. The use of heterologous antilymphoid agents in canine renal and liver homotransplantation and in human renal homotransplantation. Surg. Gynecol. Obstet. 1967; 124: 301-318. 6. Kriett JM, Kaye MP. The registry of the international society for heart and lung transplantation; eighth official report-1991. J Heart Lung Transplant 1991; 10: 491-498. 7. Starzl TE, Demetris AJ. Liver transplantation. Chicago: Year book medical publisher Inc, 1990 8. Morris PJ. Results of renal transplantation. In: Morris PJ, ed. Kidney Transplantation. Fourth ed. Saunders, Philadelphia, 1994: 504-523. 9. Cohen J, Hopkin J, Kurtz J. Infectious complications after renal transplantation. In: Morris PJ, ed. Kidney transplantation. third ed. W. B. Saunders Company, Philadelphia, 1988: 10. Sheil AGR. Complications of immunosuppression in renal allograft recipient: Malignancy. Clinical Transplantation 1991; 5: 573-579. 11. Kahan BD. Cyclosporine. N Engl J Med 1989; 321: 1735-1729. 12. Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature 1953; 172: 603-606. 13. Murray JE, Merrill JP, Harrison JH. Renal homotransplantation in identical twins. Surg Forum 1955; 6: 432. 14. Murray JE, Merrill JP, Dammin GJ, Dealy Jr JB, Walter CW, Brooke MS, Wilson PE. Study of transplantation immunity after total body irradiation: clinical and experimental investigation. Surgery 1960; 48: 272-284. 15. Calne RY. The rejection of renal homografts: inhibition in dogs by 6-mercaptopurine. Lancet 1960; 1: 417-418. 16. Murray JE, Merrill JP, Dammin GJ, Dealy Jr JB, Alexandre GW, Harrison JH. Kidney transplantation in modified recipients. Ann Surg 1962; 156: 337-355. 17. Slapak M, Geoghagen T, Digard N, Ahmed K, Sharman V, Crockett R. The use of low-dose cyclosporine in combination with azathioprine and steroids in renal transplantation. Transplant Proc 1985; 17: 1222-1226. 18. Brinker KR, Dickerman RM, Gonwa TW, Hull AR, Langley JW, Long DY, Nesser DA, Trevino G, Velez RL, Vergne-Marini PJ. A randomized trial comparing double-drug and triple drug therapy in primary cadaveric renal transplants. Transplantation 1990; 50: 43-49. 19. Persson H, Anderson C, Lundgren C, Albrechtsen D, Gabel H, Frodin L, Fehrman I, Flatmark A, Brynger H. Improved renal graft function in triple-drug treatment with low-dose cyclosporine. Transplant Proc 1987; 19: 3586-3588. 20. Borel JF. Cumulative study of in vitro and in vivo drug effects on cell mediated cytotoxicity. Immunology 1976; 31: 631-641. 21. Mathe G, Jammet H, Pendic B. Transfusion et greffes de molle osseuse homolgue chez des humains irrdiessa hautes dose accidentellement. Rev Fr Etundes Clin Biol 1959; 4: 226-233. 22. Starzl TE, Marchioro TL, von Kaulla KN. Homotransplantation of the liver in humans. Surg Gynecol Obstet 1963; 117: 659-676. 23. Barnard CN. The operation. A human cardiac transplantation in man. South Afr Med J 1967; 41: 1271-1274. 24. Thomson AW. FK506 How much potential? Immunol Today 1989; 10: 6-9. 25. Thomson AW. FK506 enters the clinic. Immunol Today 1990; 11: 35-36. 26. Natsumeda Y, Ohno S, Kawasaki H, Konno Y, Weber G, Suzuki K. Two distinct cDNAs for human IMP dehydrogenase. J Biol Chem 1990; 265: 5292-5295. 27. Morris RE. Rapamycin: FK506's twin or distant cousin? Immunol Today 1991; 12: 137-140. 28. Takeuchi T, Inuma H, Kunimoto S, Masuda T, Ishizuka M, Takeuchi M, Hamada M, Naganawa H, Kondo S, Umezawa H. A new antitumor antibiotic, spergualin: isolation and antitumor activity. J Antibiot 1981; 34: 1619-1621. 29. Kuchle CCA, Thoenes GH, Langer KH, Schorlemmer HU, Bartlett RR, Schleyerbach R. Prevention of kidney and skin graft rejection in rats by leflunomide, a new immunomodulating agent. Transplant Proc 1991; 23: 1083-1086. 30. Cramer DV, Chapman FA, Jaffee BD, Jones EA, Knoop M, Hreha-Eiras G, Makowka L. The effect of a new immunosuppressive drug, brequinar sodium, on heart, liver and kidney allograft rejection in the rat. Transplantation 1992; 53: 303-314. 31. Cosimi AB, Colvin RB, Burton RC, Rubin RH, Goldstein G, Kung DC, Hansen WP, Delmonico FL, Russell PS. Use of monoclonal antibodies to T-cell subsets for immunologic monitoring and treatment in recipients of renal allografts. N Engl J Med 1981; 305: 308-314. 32. London NJ, Farmery SM, Will EJ, Davison AM, Lodge JPA. Risk of neoplasia in renal transplant patients. Lancet 1995; 346: 403-406. 33. Gaya SBM, Rees AJ, Lechler RI, Williams G, Mason PD. Malignant disease in patients with long-term renal transplant. Transplantation 1995; 59: 1705-1709. 34. Azuma H, Tilney NL. Chronic graft rejection. Curr Opin Immunol 1994; 6: 770-776. 35. McDevitt HO, Tyan ML. Genetic control of the antibody response in inbred mice. Transfer of response by spleen cells and linkage to the major histocompatibility (H-2) locus. J Exp Med 1968; 128: 1-12. 36. Zinkernagel RM, Doherty PC. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 1974; 248: 701-702. 37. Madden DR. The three-dimensional structure of peptide-MHC complex. Annu Rev Immunol 1995; 13: 587-622. 38. Rosenthal AS, Shevach EM. Function of macrophages in antigen recognition by guinea pig T lymphocytes. I. Requirement for histocompatible macrophages and lymphocytes. J Exp Med 1973; 138: 1194-1212. 39. Chien Y-h, Davis MM. How ab T-cell receptors 'see' peptide/MHC complexes. Immunol Today 1993; 14: 597-601. 40. Gibson TM, Medawar PB. The fate of skin homografts in man. J Aat 1943; 77: 299. 41. Medawar PB. A second study of the behavior and fate of skin homografts in rabbits. J Anat 1945; 79: 157. 42. Rygaard J. Skin grafts in nude mice. Acta Pathol Microbiol Scand 1974; 82: 93-104. 43. Hall BM, Dorsch S, Roser B. The cellular basis of allograft rejection in vivo. J Exp Med 1978; 148: 878-889. 44. Rolstad B, Ford WL. Immune responses of rats deficient in thymus-derived lymphocytes to strong transplantation antigens. Transplantation 1974; 17: 405-415. 45. Hayry P, Defendi V. Mixed lymphocytes cultures produce effector cells: model in vitro for allograft rejection. Science 1970; 168: 133-135. 46. Hodes RJ, Svedmyr EAJ. Specific cytotoxicity of H-2 incompatible mouse lymphocytes following mixed culture in vitro. Transplantation 1970; 9: 470-477. 47. Engers HD, Glasebrook AL, Sorenson GD. Allogeneic tumor rejection induced by the intravenous injection of Lyt-2+ cytolytic T lymphocytes clones. J Exp Med 1982; 156: 1280-1285. 48. Tyler JD, Galli SJ, Snider ME, Dvorak AM, Steinmuller D. Cloned LYT-2+ cytolytic T lymphocytes destroy allogeneic tissue in vivo. J Exp Med 1984; 159: 234-243. 49. Rosenberg AS, Singer A. Evidence that the effector mechanism of skin allograft rejection is antigen-specific. Proc Natl Acad Sci USA 1988; 85: 7739-7742. 50. Mintz B, Silbers WK. Histocompatibility antigens on melanoblasts and hair follicule cells. Transplantation 1970; 9: 497-505. 51. Liew FY, Simpson E. Delayed-type hypersensitivity responses to H-Y: characterization and mapping of Ir genes. Immunogenetics 1980; 11: 255-266. 52. Loveland BE, Hogarth PM, Ceredig R, McKenzie IF. Delayed type hypersensitivity and allograft rejection in the mouse: correlation of effector cell phenotype. J Exp Med 1981; 153: 1044-1057. 53. Dallman MJ, Mason DW, Webb M. Induction of antigens on murine epidermal cells during the rejection of skin allograft. Eur J Immunol 1982; 12: 511-518. 54. Gurley KE, Lowry RP, Clarke-Forbes RD. Immune mechanisms in organ allografts rejection: II. T helper cells, delayed type hypersensitivity and rejection of renal allografts. Transplantation 1983; 36: 401-405. 55. Hall BM, DeSaxe I, Dorsch SE. The cellular basis of allograft rejection in vivo: restoration of first set rejection of heart grafts by T helper cells in irradiated rats. Transplantation 1983; 36: 700-705. 56. Lowry RP, Guley KE, Clarke-Forbes RD. Immune mechanisms in organ allograft rejection. Transplantation 1983; 36: 391-401. 57. Sprent J, Schaeffer M, Lo D. Properties of purified T cell subsets II. In vivo class I vs class II H-2 differences. J Exp Med 1986; 163: 998-1011. 58. Rosenberg AS, Mizuochi T, Singer A. Analysis of T-cell subsets in rejection of Kb mutant skin allografts differing at class I MHC. Nature 1986; 322: 829-831. 59. Rosenberg AS, Mizuochi T, Sharrow SO, Singer A. Phenotype of specificity and function of T cell subsets and T cell interactions involves in skin allograft rejection. J Exp Med 1987; 165: 1296-1315. 60. Galili U. Interaction of the natural anti-Gal antibody with a-galactosyl epitopes: a major obstacle for xenotransplantation in humans. Immunol Today 1993; 14: 480-482. 61. Kaufman CL, Gaines BA, Ildstad ST. Xenotransplantation. Annu Rev Immunol 1995; 13: 339-367. 62. Morris PJ, Ting A. Studies of HLA-DR with relevance to renal transplantation. Immunol Rev 1982; 66: 103-131. 63. Baldwin WM, Pruitt SK, Sanfilippo F. Alloantibodies: basic and clinical concepts. Transplant Rev 1991; 5: 100. 64. Mason DW, Morris PJ. Inhibition of the accumulation, in rat kidney allografts, of specific--but not nonspecific--cytotoxic cells by cyclosporine. Transplantation 1984; 37: 46-51. 65. Bradley JA, Mason DW, Morris PJ. Evidence that rat renal allografts are rejected by cytotoxic T cells and not by nonspecific effectors. Transplantation 1985; 39: 169-175. 66. Armstrong HE, Bolton EM, McMillan I, C. SS, A. BJ. Prolonged survival of actively enhanced rat renal allografts despite accelerated cellular infiltration and rapid induction of both class I and class II MHC antigens. J Exp Med 1987; 165: 891-907. 67. Murphy WJ, Kumar V, Bennett M. Acute rejection of murine bone marrow allografts by natural killer cells and T cells. Differences in kinetics and target antigens recognized. J Exp Med 1987; 166: 1499-1509. 68. Murphy W, Kumar V, Bennett M. Rejection of bone marrow allografts by mice with severe combined immune deficiency (SCID): evidence that NK cells can mediate the specificity of morrow graft rejection. J Exp Med 1987; 165: 1212-1217. 69. Grusby MJ, Johnson RS, Papaioannou V, Glimcher LH. Depletion of CD4+ T cells in major histocompatibility class II-deficient mice. Science 1991; 253: 1417-1420. 70. Grusby M, Auchincloss H, Lee R, Johnson R, Spencer J, Zijlstra M, Jaenisch R, Papaioannou V, Glimcher L. Mice lacking major histocompatibility complex class I and class II molecules. Proc Natl Acad Sci USA 1993; 90: 3913-3917. 71. Shoskes DA, Wood KJ. Indirect presentation of MHC antigens in transplantation. Immunol Today 1994; 15: 32-38. 72. Lechler RI, Batchelor JR. Restoration of immunogenicity to passenger cell-depleted kidney allografts by the addition of donor strain dendritic cells. J Exp Med 1982; 155: 31-41. 73. Sherwood RA, Brent L, Rayfield LS. Presentation of alloantigens by host cells. Eur J Immunol 1986; 16: 569-574. 74. Benichou G, Takizawa PA, Olson CA, McMillan M, Serarz EF. Donor major histocompatibility complex (MHC) peptides are presented by recipient MHC molecules during graft rejection. J Exp Med 1992; 175: 305-308. 75. Auchincloss HJ, Lee R, Shea S, Markowitz JS, Grusby MJ, Glimcher LH. The role of "indirect" recognition in initiating recognition of skin grafts from major histocompatibility complex class II-deficient mice. Proc Natl Acad Sci USA 1993; 90: 3373-3377. 76. Lee RS, Grusby MJ, Winn HJ, Auchincloss HJ. Indirect recognition by helper cells can induce donor-specific cytotoxic T lymphocytes in vivo. J Exp Med 1994; 179: 865-872. 77. Braun MY, McCormack A, Webb G, Batchelor JR. Mediation of acute but not chronic rejection of MHC-incompatible rat kidney grafts by alloreactive CD4 T cells activated by the direct pathway of sensitization. Transplantation 1993; 55: 177-182. 78. Snell GD. Methods for the study of histocompatibility genes. J. Genet. 1948; 49: 87-108. 79. Ting A, Morris PJ. Matching for B-cell antigens of the HLA-DR (D-related) series in cadaver renal transplantation. Lancet 1978; 1: 575-577. 80. Peugh WN, Superina RA, Wood KJ, Morris PJ. The role of H-2 and non-H-2 antigens and genes in the rejection of murine cardiac allografts. Immunogenetics 1986; 23: 30-37. 81. Perreault C, Roy DC, Fortin C. Immunodominant minor histocompatibility antigens: the major ones. Immunol Today 1998; 19: 69-74. 82. Eichwald EJ, Silmser CR. Untitled. Transplant Bull 1955; 2: 148-149. 83. Bailey DW. Allelic forms of a gene controlling the female immune response to the male antigen in mice. Transplantation 1971; 11: 426-428. 84. Bailey DW, Hoste J. A gene governing the female immune response to the male antigen in mice. Transplantation 1971; 11: 404-407. 85. Tourne S, Miyazaki T, Wolf P, Ploegh H, Benoist C, Mathis D. Functionality of major histocompatibility complex class II molecules in mice doubly deficient for invariant chain and H-2M complexes. Proc Natl Acad Sci USA 1997; 94: 9255-9260. 86. Chang CH, Guerder S, Hong SC, van Ewijk W, Flavell RA. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity 1996; 4: 167-178. 87. Karlsson L, Peterson PA. The alpha chain gene of H-2O has an unexpected location in the major histocompatibility complex. J Exp Med 1992; 176: 477-483. 88. Owen RD. Immunogenetic consequences of vascular anastomoses between bovine twins. Science 1945; 102: 400-401. 89. Burnett FM. A modification of Jerne's theory of antibody production using the concept of clonal selection. Aust. J. Sci. 1957; 20: 67-69. 90. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994; 12: 991-1045. 91. Nikolic-Zugic J. Phenotypic and functional stages in the intrathymic development of ab T cells. Immunol Today 1991; 12: 65-79. 92. Kisielow P, Bluthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature 1988; 333: 742-746. 93. Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell 1987; 49: 273-280. 94. MacDonald HR, Schneider R, Lees RK, Howe RC, Acha-Orbea H, Festenstein H, Zinkernagel RM, Hengartner H. T-cell receptor Vb use predicts reactivity and tolerance to Mlsa-encoded antigens. Nature 1988; 332: 40-45. 95. Sha WC, Nelson CA, Newberry RD, Kranz CM, Russell JH, Loh DY. Positive and negative selection of an antigen receptor on T cells in transgenic mice. Nature 1988; 336: 73-76. 96. Marrack P, Kappler J. The T cell repertoire for antigen. Immunol Today 1988; 9: 308-315. 97. Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: A costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol 1989; 7: 445-480. 98. Burkly LC, Lo D, Kanahawa O, Brinster RL, Flavell RA. T-cell tolerance by clonal anergy in transgenic mice with nonlymphoid expression of MHC class II I-E. Nature 1989; 342: 564-566. 99. Morahan G, Allison J, Miller JFAP. Tolerance of class I histocompatibility antigens expressed extrathymically. Nature 1989; 339: 622-624. 100. Ashton-Rickardt PG, Bandeira A, Delaney JR, Kaer LV, Pircher H-P, Zinkernage RM, Tonegawa S. Evidence for a differential avidity model of T cell selection in the thymus. Cell 1994; 76: 651-663. 101. Sebzda E, Wallace VA, Mayer J, Yeung RSM, Mak TW, Ohashi PS. Positive and negative thymocyte selection induced by different concentrations of a single peptide. Science 1994; 263: 1615-1618. 102. Rammensee H-G, Kroschewski R, Frangoulis B. Clonal anergy induced in mature Vb6+ T lymphocytes on immunizing Mls-1b mice with Mls-1a expressing cells. Nature 1989; 339: 541-544. 103. Bretscher P, Cohn M. A theory of self-nonself discrimination. Science 1970; 169: 1042-1049. 104. Lafferty KJ, Prowse SJ, Simeonovic CJ. Immunobiology of tissue transplantation: a return to the passenger leukocyte concept. Annu Rev Immunol 1983; 1: 143-173. 105. Lafferty KJ, Cooley MA, Woolnough J, Walker KZ. Thyroid allograft immunogenicity is reduced after a period in organ culture. Science 1975; 188: 259-261. 106. Lamb JR, Feldmann M. Essential requirement for major histocompatibility complex recognition in T-cell tolerance induction. Nature 1984; 308: 72-74. 107. Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med 1987; 165: 302-319. 108. Quill H, Schwartz RH. Stimulation of normal inducer T cell clones with antigen presented by purified Ia molecules in planar lipid membranes: specific induction of a long-lived state of proliferative nonresponsiveness. J Immunol 1987; 138: 3704-3712. 109. Gaspari AA, Jenkins MK, Katz SI. Class II MHC-bearing keratinocytes induce antigen-specific unresponsiveness in hapten-specific Th1 clones. J Immunol 1988; 141: 2216-2220. 110. Markmann J, Lo D, Naji A, Palmiter RD, Brinster RL, Heber-Katz E. Antigen presenting function of class II MHC expressing pancreatic beta cells. Nature 1988; 336: 476-479. 111. Cho EA, Riley MP, Sillman AL, Quill H. Altered protein tyrosine phosphorylation in anergic Th1 cells. J Immunol 1993; 151: 20-28. 112. Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992; 356: 607-609. 113. Isobe M, Yagita H, Okumura K, Ihara A. Specific acceptance of cardiac allograft after treatment with antibodies to ICAM-1 and LFA-1. Science 1992; 255: 1125-1127. 114. Noelle RJ. CD40 and its ligand in host defence. Immunity 1996; 4: 415-419. 115. Nemazee DA, Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature 1989; 337: 562-566. 116. Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, Trent RJ, Basten A. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 1988; 334: 676-682. 117. Goodnow CC, Crosbie J, Jorgensen H, Brink RA, Basten A. Induction of self-tolerance in mature peripheral B lymphocytes. Nature 1989; 342: 385-391. 118. Goodnow CC, Brink R, Adams E. Breakdown of self-tolerance in anergic B lymphocytes. Nature 1991; 352: 532-536. 119. Ramsdell F, Fowlkes BJ. Maintenance of in vivo tolerance by persistence of antigen. Science 1992; 257: 1130-1134. 120. Hamano K, Rawsthorne MA, Bushell AR, Morris PJ, Wood KJ. Evidence that the continued presence of the organ graft and not peripheral donor microchimerism is essential for maintenance of tolerance to alloantigen in vivo in anti-CD4 treated recipients. Transplantation 1996; 62: 856-860. 121. Qin S, Wise M, Cobbold SP, Leong L, Kong Y-CM, Parnes JR, Waldmann H. Induction of tolerance in peripheral T cells with monoclonal antibodies. Eur J Immunol 1990; 20: 2737-2745. 122. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, Waldmann H. "Infectious" transplantation tolerance. Science 1993; 259: 974-977. 123. Roser BJ. Cellular mechanisms in neonatal and adult tolerance. Immunol Rev 1989; 107: 179-202. 124. Streilein JW, Gruchalla RS. Analysis of neonatally induced tolerance of H-2 alloantigens: I. Adoptive transfer indicates that tolerance of class I and class II antigens is maintained by distinct mechanisms. Immunogenetics 1981; 12: 161-173. 125. Takeuchi T, Lowry RP, Konieczny B. Heart allografts in murine systems: the differential activation of Th2-like effector cells in peripheral tolerance. Transplantation 1992; 53: 1281-1294. 126. Hall BM, Pearce NW, Gurley KE, Dorsch SE. Specific unresponsiveness in rats with prolonged cardiac allografts survival after treatment with cyclosporine: III. Further Characterization of the CD4+ suppressor cell and its mechanisms of action. J Exp Med 1990; 171: 141-157. 127. Scully R, Qin S, Cobbold S, Waldmann H. Mechanisms in CD4 antibody-mediated transplantation tolerance: kinetics of induction, antigen dependency and role of regulatory T cells. Eur J Immunol 1994; 24: 2383-2392. 128. Dallman MJ, Wood KJ, Morris PJ. Specific cytotoxic T cells are found in the nonrejected kidneys of blood-transfused rats. J Exp Med 1987; 165: 566-571. 129. Streilein JW. Overview---neonatal tolerance of H-2 alloantigens: procuring graft acceptance "the old-fashioned way". Transplantation 1991; 52: 1-10. 130. Fowell D, Mason D. Evidence that the T cell repertoire of normal rats contains cells with the potential to cause diabetes. Characterization of the CD4+ T cell subset that inhibits this autoimmune potential. J Exp Med 1993; 177: 627-636. 131. Morrissey PJ, Charrier K, Braddy S, Liggitt D, Watson JD. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J Exp Med 1993; 178: 237-244. 132. Powrie F, Correa-Oliveira R, Mauze S, Coffman RL. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance protective and pathogenic cell-mediated immunity. J Exp Med 1994; 179: 589-600. 133. Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell response and prevent colitis. Nature 1997; 389: 737-742. 134. Mosmann TR, Coffman RL. Th1 and Th2 cells. Different pattern of lymphokine secretion lead to different functional properties. Annu Rev Immunol 1989; 7: 145-173. 135. Sad S, Mosmann TR. Single IL-2-secreting precursor CD4 T cell can develop into either Th1 or Th2 cytokine secretion phenotype. J Immunol 1994; 153: 3514-3522. 136. Takahashi T, Mafune K, Maki T. Cloning of self-major histocompatibility complex antigen-specific suppressor cells from adult bone marrow. J Exp Med 1990; 172: 901-909. 137. Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today 1995; 16: 34-38. 138. Chen N, Field EH. Enhanced type 2 and diminished type 1 cytokines in neonatal tolerance. Transplantation 1995; 59: 933-941. 139. Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 1994; 265: 1237-1240. 140. Melamed D, Friedman A. In vivo tolerization of Th1 lymphocytes following a single feeding with ovalbumin: anergy in the absence of suppression. Eur J Immunol 1994; 24: 1974-1981. 141. Powell TJ, Streilein JW. Neonatal tolerance induction by class II alloantigens activates IL-4-secreting, tolerogen-responsive T cells. J Immunol 1990; 144: 854-859. 142. Chen N, Gao Q, Field EH. Expansion of memory Th2 cells over Th1 cells in neonatal primed mice. Transplantation 1995; 60: 1187-1193. 143. Chen N, Gao Q, Field EH. Prevention of Th1 responses is critical for tolerance. Transplantation 1996; 61: 1076-1183. 144. Gao Q, Chen N, Rouse TM, Field EH. The role of IL-4 in the induction phase of allogeneic neonatal tolerance. Transplantation 1996; 62: 1847. 145. Nickerson P, Steurer W, Steinger J, Zheng X, Steele AW, Strom TB. Cytokines and the Th1/Th2 paradigm in transplantation. Curr Opin Immunol 1994; 6: 757-764. 146. Saoudi A, Simmonds S, Huitinga I, Mason D. Prevention of experimental allergic encephalomyelitis in rats by targeting autoantigen to B cells: Evidence that the protective mechanism depends on changes in the cytokine response and migratory properties of the autoantigen-specific T cells. J Exp Med 1995; 182: 335-344. 147. Fowler DH, Kurasawa K, Smith R, Eckhaus MA, Gress R. Donor CD4-enriched cells of Th2 cytokine phenotype regulate graft-versus-host disease without impairing allogeneic engraftment in sublethally irradiated mice. Blood 1994; 84: 3540-3549. 148. Maeda H, Takata M, Takahashi S, Ogoshi S, Fujimoto S. Adoptive transfer of a Th2-like cell line prolongs MHC class II antigen disparate skin allograft survival in the mouse. Int Immunol 1994; 6: 855-862. 149. Bishop GA, Sun J, Sheil AGR, McCaughan GW. High-dose/activation-associated tolerance. Transplantation 1997; 64: 1377-1382. 150. Ohashi PS, Oehen S, Buerki K, Pircher H, Ohashi CT, Odermatt B, Malissen B, Zinkernagel RM, Hengartner H. Ablation of "tolerance" and induction of diabetes by virus infection in viral antigen transgenic mice. Cell 1991; 65: 305-317. 151. Allison J, Campbell IL, Morahan G, Miller JFAP. Diabetes in transgenic mice resulting from over-expression of class I histocompatibility molecules in pancreatic beta cells. Nature 1988; 333: 529-533. 152. Heath WR, Allison J, Hoffmann MW, Schonrich G, Hammerling G, Arnold B, Miller JF. Autoimmune diabetes as a consequence of locally produced interleukin-2. Nature 1992; 359: 547-549. 153. Mosier DE. A requirement for two cell types for antibody formation in vitro. Science 1967; 158: 1573-1575. 154. Oppenheim JJ, Leventhal BG, Hersh EM. The transformation of column-purified lymphocytes with nonspecific and specific antigenic stimuli. J Immunol 1968; 101: 262-270. 155. Inaba K, Steinman RM. Resting and sensitized T lymphocytes exhibit distinct stimulatory(antigen-presenting cell) requirements for growth and lymphokine release. J Exp Med 1984; 160: 1717-1735. 156. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med 1973; 137: 1142-1162. 157. Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 1991; 9: 271-296. 158. Ingulli E, Mondino A, Khoruts A, Jenkins MK. In vivo detection of dendritic cell antigen presentation to CD4+ T cells. J Exp Med 1997; 185: 2133-2141. 159. Kudo S, Matsuno K, Ezaki T, Ogawa M. A novel migration pathway for rat dendritic cells from the blood: hepatic sinusoids-lymph translocation. J Exp Med 1997; 185: 777-784. 160. Watts C. Inside the gearbox of the dendritic cell. Nature 1997; 388: 724-725. 161. Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Mirza A, Inaba K, Steinman RM, Mellman I. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature 1997; 388: 787-792. 162. Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature 1997; 388: 782-787. 163. Austyn JM, Weinstein DE, Steinman RM. Clustering with dendritic cells precedes and is essential for T-cell proliferation in a mitogenesis model. Immunology 1988; 63: 691-696. 164. Flechner ER, Freudenthal PS, Kaplar G, Steinman RM. Antigen-specific T lymphocytes efficiently cluster with dendritic cells in the human primary mixed-leukocyte reaction. Cell Immunol 1988; 111: 183-195. 165. Inaba K, Steinman RM. Accessory cell-T lymphocyte interactions. J Exp Med 1986; 163: 247-261. 166. Chesnut RW, Grey HM. Studies on the capacity of B cells to serve as antigen-presenting cells. J Immunol 1981; 126: 1075-1079. 167. Chesnut RW, Colon SM, Grey HM. Antigen presentation by normal B cells, B cell tumors, and macrophages: functional and biochemical comparison. J Immunol 1982; 128: 1767-1768. 168. Kakiuchi T, Chesnut RW, Grey HM. B cells as antigen-presenting cells: the requirement for B cell activation. J Immunol 1983; 109-114. 169. Ashwell JD, DeFranco AL, Paul WE, Schwartz RH. Antigen presentation by resting B cells: Radiosensitivity of the antigen-presentation function and two distinct pathways of T cell activation. J Exp Med 1984; 159: 881-905. 170. Krieger JI, Grammer SF, Grey HM, Chesnut RW. Antigen presentation by splenic B cells: Resting B cells are ineffective, whereas activated B cells are effective accessory cells for T cell responses. J Immunol 1985; 135: 2937-2945. 171. Ashwell JD. Are B lymphocytes the principal antigen-presenting cells in vivo. J Immunol 1988; 140: 3697-3700. 172. Ashwell JD, Jenkins MK, Schwartz RH. Effect of gamma radiation on resting B lymphocytes II. Functional characterization of the antigen-presentation defect. J Immunol 1988; 141: 2536-2544. 173. Eynon EE, Parker DC. Do small B cells induce tolerance. Transplant Proc 1991; 23: 729-730. 174. Eynon EE, Parker DC. Small B cells as antigen-presenting cells in the induction of tolerance to soluble protein antigens. J Exp Med 1992; 175: 131-138. 175. Eynon EE, Parker DC. Parameters of tolerance induction by antigen targeted to B lymphocytes. J Immunol 1993; 151: 2958-64. 176. Lacy PE, Finke EH, Janey CG, Davie JM. Prolongation of islet xenograft survival by in vitro culture of rat megaislets in 95%O2. Transplantation 1982; 33: 588-592. 177. Fabre JW, Morris PJ. The role of passenger leukocytes in the rejection of renal allografts in the rat. Transplantation 1973; 15: 631-633. 178. Lafferty KJ, Bootes A, Dart G, Talmage DW. Effect of organ culture on the survival of thyroid allografts in mice. Transplantation 1976; 22: 138-149. 179. Steinmuller D, Hart EA. Passenger leukocytes and induction of allograft immunity. Transplant Proc 1971; 3: 673-675. 180. Stuart FP, Bastien E, Holter A, Fitch FW, Elkins WL. Role of passenger leukocytes in the rejection of renal allografts. Transplant Proc 1971; 3: 461-464. 181. Faustman D, Lacy P, Hauptfeld V. Prevention of allograft rejection by immunization with donor blood depleted of Ia-bearing cells. Science 1982; 217: 157-158. 182. Lau H, Reemtsma K, Hardy MA. pancreatic islet allograft prolongation by donor-specific blood transfusions treated with ultraviolet irradiation. Science 1983; 221: 754-756. 183. Ryan JJ, Gress RE, Hathcock KS, Hodes RJ. Recognition and response to alloantigens in vivo II. Priming with accessory cell-depleted donor allogeneic splenocytes: induction of specific unresponsiveness to foreign major histocompatibility complex determinants. J Immunol 1984; 133: 2343-2350. 184. Hori S, Sato S, Kitagawa S, Azuma T, Kokudo S, Hamaoka T, Fujiwara H. Tolerance induction of allo-class II H-2 antigen-reactive L3T4+ helper T cells and prolonged survival of the corresponding class II H-2-disparate skin graft. J Immunol 1989; 143: 1447-1452. 185. Fuchs EJ, Matzinger P. B cells turn off virgin but not memory T cells. Science 1992; 258: 1156-1159. 186. Dossetor JB, MacKinnon KJ, Gault MH, MacLean LD. Cadaver kidney transplants. Transplantation 1967; 5: 844-853. 187. Morris PJ, Ting A, Stocker JW. Leukocyte antigens in renal transplantation. The paradox of blood transfusion in renal transplantation. Med J Aust 1968; 2: 1088-1090. 188. Opelz G, Vanrenterghem Y, Kirste G, Gray DW, Horsburgh T, Lachance JG, Largiader G, Lange H, Vujaklija-Stipanovic K, Alvarez-Grande J, Schott W, Hoyer J, Schnuelle P, Descoeudres C, Ruder H, Wujciak T, Schwarz V. Prospective evaluation of pretransplant blood transfusions in cadaver kidney recipients. Transplantation 1997; 63: 964-967. 189. Marquet RL, Heystek GA, Tenbergen WJ. Specific inhibition of organ allograft rejection by donor blood. Transplant Proc 1971; 3: 708-710. 190. Fabre JW, Morris PJ. The effect of donor strain blood pretreatment on renal allograft rejection in rat. Transplantation 1972; 14: 608-617. 191. Chen C, Nabavi N. In vitro induction of T cell anergy by blocking B7 and early T cell costimulatory molecule ETC-1/B7-2. Immunity 1994; 1: 147-154. 192. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996; 14: 233-258. 193. Aruffo A, Seed B. Molecular cloning of a CD28 cDNA by a high efficiency COS cell expression system. Proc Natl Acad Sci USA 1987; 84: 8573-8577. 194. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, Golstein P. A new member of the immunoglobulin superfamily---CTLA-4. Nature 1987; 328: 267-270. 195. Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol 1993; 11: 191-212. 196. June CH, A. LJ, Linsley PS, Thompson CB. Role of the CD28 receptor in T-cell activation. Immunol Today 1990; 11: 211-216. 197. Gross JA, John TS, Allison JP. The murine homologue of the T lymphocyte antigen D28. J Immunol 1990; 144: 3201-3210. 198. Gross JA, Callas E, Allison JP. Identification and distribution of the costimulatory receptor CD28 in the mouse. J Immunol 1992; 149: 380-388. 199. Turka LA, Ledbetter JA, Lee K, June CH, Thompson CB. CD28 is an inducible T cell surface antigen that transduces a proliferative signal in CD3+ mature thymocytes. J Immunol 1990; 144: 1646-1653. 200. Eck SC, Chang D, Wells AD, Turka LA. Differential down-regulation of CD28 by B7-1 and B7-2 engagement. Transplantation 1997; 64: 1497-1488. 201. Linsley PS, Greene JL, Tan P, Bradshaw J, Ledbetter JA, Anasetti C, Damle NK. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med 1992; 176: 1595-1604. 202. Freeman GJ, Lombard DB, Gimmi CD, Brod SA, Lee K, Laning JC, Hafler DA, Dorf ME, Gray GS, Reiser H. CTLA-4 and CD28 mRNA are coexpressed in most T cells after activation. Expression of CTLA-4 and CD28 mRNA does not correlate with the pattern of lymphokine production. J Immunol 1992; 149: 3795-3801. 203. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompsom CB, Bluesone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994; 1: 405-413. 204. Damle NK, Klussman K, Leytze G, Myrdal S, Aruffo A, Ledbetter JA, Linsley PS. Costimulation of T lymphocytes with integrin ligands intercellular adhesion molecule-1 or vascular cell adhesion molecule-1 induces functional expression of CTLA-4, a second receptor for B7. J Immunol 1994; 152: 2686-2697. 205. Green JM, Noel PJ, Sperling AI, Walunas TL, Gray GS, Bluestone JA, Thompson CB. Absence of B7-dependent responses in CD28-deficient mice. Immunity 1994; 1: 501-508. 206. Parijs LV, Ibraghimov A, Abbas AK. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity 1996; 4: 321-328. 207. Sperling AI, Auger JA, Ehst BD, Rulifson IC, Thompson CB, Bluestone JA. CD28/B7 interaction delivered a unique signal to naive T cells that regulates cell survival but not early proliferation. J Immunol 1996; 157: 3909-3917. 208. Lucas PJ, Negishi I, Nakayama K, Fields LE, Loh DY. Naive CD28-deficient T cells can initiate but not sustain an in vitro antigen-specific immune response. J Immunol 1995; 154: 5757-5768. 209. Boise LH, Minn AJ, Noel PJ, June CH, Accavitti M, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xL. Immunity 1995; 3: 87-98. 210. Nakayama K-I, Nakayama K, Negishi I, Kuida K, Shinkai Y, Louie MC, Fields LE, Lucas PJ, Stewart V, Alt FW, Loh DY. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science 1993; 261: 1584-1588. 211. Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that block programmed cell death. Nature 1990; 348: 334-336. 212. Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptosis cell death. Cell 1993; 74: 597-608. 213. McArthur JG, Raulet DH. CD28-induced costimulation of T helper type 2 cells mediated by induction of responsiveness to interleukin 4. J Exp Med 1993; 178: 1645-1653. 214. Thompson CB, Lindsten T, Ledbetter JA, Kunkel SL, Young HA, Emerson SG, Leiden JM, June CH. CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc Natl Acad Sci USA 1989; 86: 1333-1337. 215. Seder RA, Germain RN, Linsley PS, Paul WE. CD28-mediated costimulation of interleukin 2 (IL-2) production plays a critical role in T cell priming for IL-4 and interferon gamma production. J Exp Med 1994; 179: 299-304. 216. King CL, Stupi RJ, Craighead N, June CH, Thyphronitis G. CD28 activation promotes Th2 subset differentiation by human CD4+ cells. Eur J Immunol 1995; 25: 587-595. 217. van der Pouw-Kraan T, van Kooten C, Rensink I, Aarden L. Interleukin (IL)-4 production by human T cells: differential regulation of IL-4 vs. IL-2 production. Eur J Immunol 1992; 22: 1237-1241. 218. Shahiniam A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993; 261: 609-612. 219. Lenschow DJ, Herold KC, Rhee L, Patel B, Koons A, Qin H-Y, Fuchs E, Singh B, Thompson CB, Bluestone JA. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity 1996; 5: 285-293. 220. Sayegh MH, Akalin E, Hancock WW, Russell ME, Carpenter CB, Linsley PS, Turka LA. CD28-B7 blockade after alloantigenic challenge in vivo inhibits Th1 cytokines but spares Th2. J Exp Med 1995; 181: 1869-1874. 221. Khoury SJ, Akalin E, Chandraker A, Turka LA, Linsley PS, Sayegh MH, Hancock WW. CD28-B7 costimulatory blockade by CTLA4Ig prevents actively induced experimental autoimmune encephalomyelitis and inhibits Th1 but spares Th2 cytokines in the central nervous system. J Immunol 1995; 155: 4521-4524. 222. Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: The alternative approaches. Annu Rev Immunol 1997; 15: 297-322. 223. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995; 182: 459-465. 224. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995; 3: 541-547. 225. Waterhouse P, Penninger JM, Timms E, Waleham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995; 270: 985-988. 226. Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity 1997; 7: 885-895. 227. Allison JP, Krummel MF. The yin and yang of T cell costimulation. Science 1995; 270: 932-933. 228. Thompson CB, Allison JP. The emerging role of CTLA-4 as an immune attenuator. Immunity 1997; 7: 445-450. 229. Liu Y. Is CTLA-4 a negative regulator for T-cell activation? Immunol Today 1997; 18: 569-572. 230. Yokochi T, Holly RD, Clark EA. B lymphoblast antigen (BB-1) expressed on Epstein-Barr virus-activated B cell blasts, B lymphoblastoid cell lines, and Burkitt's lymphomas. J Immunol 1982; 128: 823-827. 231. Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci USA 1990; 87: 5031-5035. 232. Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL, Somoza C. B 70 antigen is a second ligand for CTLA-4 and CD28. Nature 1993; 366: 76-79. 233. Inaba K, Witmer-Pack M, Inaba M, Hathcock KS, Sakuta H, Azuma M, Yagita H, Okumura K, Linsley PS, Ikehara S, Muramatsu S, Hodes RJ, Steinman RM. The tissue distribution of the B7-2 costimulator in mice: abundant expression on dendritic cells in situ and during maturation in vitro. J Exp Med 1994; 180: 1849-1860. 234. Lenschow DJ, Su GHT, Zucherman LA, Navabi N, Jellis CL, Gray GS, Miller J, Bluestone JA. Expression and functional significance of an additional ligand for CTLA4. Proc Natl Acad Sci USA 1993; 90: 11054-11058. 235. Larsen CP, Ritchie SC, Pearson TC, Linsley PS, Lowry RP. Functional expression of the costimulatory molecule, B7/BB1, on murine dendritic cell populations. J Exp Med 1992; 176: 1215-1220. 236. Symington FW, Brady W, Linsley PS. Expression and function of B7 on human epidermal Langerhans cells. J Immunol 1993; 150: 1286-1295. 237. Freedman AS, Freeman GJ, Rhynhart K, Nadler LM. Selective induction of B7/BB1 on interferon gamma stimulated monocytes: a potential mechanism for amplification of T cell activation through the CD28 pathway. Cell Immunol 1991; 137: 429-437. 238. Barcy S, Wettendorff M, Leo L, Urbain J, kruger M, Ceuppens JL, de Boer M. FcR cross-linking on monocytes results in impaired T cell stimulatory capacity. Int Immunol 1995; 7: 179-189. 239. Hathcock KS, Laszlo G, Pucillo C, Linsley P, Hodes RJ. Comparative analysis of B7-1 and B7-2 costimulatory ligands: Expression and function. J Exp Med 1994; 180: 631-640. 240. Azuma M, Cayabyab M, Buck D, Phillips JH, Lanier LL. CD28 interaction with B7 costimulates primary allogeneic proliferative responses and cytotoxicity mediated by small, resting T lymphocytes. J Exp Med 1992; 175: 353-360. 241. Prabhu Das MR, Zamvil SS, Borriello F, Weiner HL, Sharpe AH, Kuchroo VK. Reciprocal expression of costimulatory molecules, B7-1 and B7-2, on murine T cells following activation. Eur J Immunol 1995; 25: 207-211. 242. Pichler WJ, Wyss-Coray T. T cells as antigen-presenting cells. Immunol Today 1994; 15: 312-315. 243. Freeman GJ, Gray GS, Gimmi CD, Lombard DB, Zhou LJ, Nadler LM. Structure, expression, and T cell costimulatory activity of the murine homologue of the human B lymphocyte activation antigen B7. J Exp Med 1991; 174: 625-631. 244. Freeman GJ, Freedman AS, Segil JM, Lee G, Whitman JF, Nadler LM. B7, a new member of the Ig superfamily with unique expression on activated and neoplastic B cells. J Immunol 1989; 143: 1714-1722. 245. Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter JA. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med 1991; 173: 721-730. 246. Gimmi CD, Freeman GJ, Gribben JG, Sugita K, Freedman AS, Morimoto C, Nadler LM. B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc Natl Acad Sci USA 1991; 88: 6575-6579. 247. Harlan DM, Hengartner H, Huang ML, Kang YH, Abe R, Moreadith RW, Pircher H, Gray GS, Ohashi PS, Freeman GJ. Mice expressing both B7-1 and viral glycoprotein on pancreatic beta cells along with glycoprotein-specific transgenic T cells develop diabetes due to a breakdown of T-lymphocyte unresponsiveness. Proc Natl Acad Sci USA 1994; 91: 3137-3141. 248. Guerder S, Meyerhoff J, Flavell R. The role of the T cell costimulator B7-1 in autoimmunity and the induction and maintenance of tolerance to peripheral antigen. Immunity 1994; 1: 155-166. 249. Wu Y, Guo Y, Liu Y. A major costimulatory molecule on antigen-presenting cells, CTLA4 ligand A, is distinct from B7. J Exp Med 1993; 178: 1789-1793. 250. Razi-Wolf Z, Galvin F, Gray G, Reiser H. Evidence for an additional ligand, distinct from B7, for the CTLA-4 receptor. Proc Natl Acad Sci USA 1993; 90: 11182-11186. 251. Hathcock KS, Laszlo G, Dickler HB, Bradshaw J, Linsley P, Hodes RJ. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science 1993; 262: 905-907. 252. Freeman GJ, Borriello F, Hodes RJ, Reiser H, Hathcock KS, Laszlo G, McKnight AJ, Kim J, Du L, Lombard DB, Gray GS, Nadler LM, Sharpe AH. Uncovering of functional alternative CTLA-4 counter-receptor in B7-deficient mice. Science 1993; 262: 907-909. 253. Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA, Lombard LA, Gray GS, Nadler LM. Cloning of B7-2: A CTLA-4 counter-receptor that costimulates human T cell proliferation. Science 1993; 262: 909-911. 254. Chen C, Gault A, Shen L, Nabavi N. Molecular cloning and expression of early T cell costimulatory molecule-1 and its characterization as B7-2 molecule. J Immunol 1994; 152: 4929-4936. 255. Freeman GJ, Borriello F, Hodes RJ, Reiser H, Gribben JG, Ng JW, Kim J, Goldberg JM, Hathcock K, Laszlo G, Lombard LA, Wang S, Gray GS, Nadler LM, Sharpe AH. Murine B7-2, an alternative CTLA4 counter-receptor that costimulates T cell proliferation and interleukin 2 production. J Exp Med 1993; 178: 2185-2192. 256. Lenschow DJ, Sperling AI, Cooke MP, Freeman G, Rhee L, Decker DC, Gray G, Nadler LM, Goodnow CC, Bluestone JA. Differential up-regulation of the B7-1 and B7-2 costimulatory molecules after Ig receptor engagement by antigen. J Immunol 1994; 153: 1990-1997. 257. Chen C, Faherty DA, Gault A, Connaughton SE, Powers GD, Godfrey DI, Nabavi N. Monoclonal antibody 2D10 recognizes a novel T cell costimulatory molecule on activated murine B lymphocytes. J Immunol 1994; 152: 2105-2114. 258. Lenschow DJ, Zeng Y, Hathcock KS, Zuckerman LA, Freeman G, Thistlethwaite JR, Gray GS, Hodes RJ, Bluestone JA. Inhibition of transplant rejection following treatment with anti-B7-2 and anti-B7-1 antibodies. Transplantation 1995; 60: 1171-1178. 259. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994; 1: 793-801. 260. Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathway: application to autoimmune disease therapy. Cell 1995; 80: 707-718. 261. Freeman GJ, Boussiotis VA, Anumanthan A, Bernstein GM, Ke X-Y, Rennert PD, Gray GS, Gribben JG, Nadler LM. B7-1 and B7-2 do not deliver identical costimulatory signals, since B7-2 but not B7-1 preferentially costimulates the initial production of IL-4. Immunity 1995; 2: 523-532. 262. Clark EA. CD40: a cytokine receptor in search of a ligand. Tissue Antigens 1990; 36: 33-36. 263. Alderson MR, Armitage RJ, Tough TW, Strockbine L, Fanslow WC, Spriggs MK. CD40 expression by human monocytes: Regulation by cytokines and activation of monocytes by the ligand for CD40. J Exp Med 1993; 178: 669-674. 264. Caux C, Massacrier C, Vanbervliet B, Dubois B, van Kooten C, Durand I, Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J Exp Med 1994; 180: 1263-1272. 265. Karmann K, Hughes CCW, Schechner J, Fanslow WC, Pober JS. CD40 on human endothelial cells: Inducibility by cytokines and functional regulation of adhesion molecule expression. Proc Natl Acad Sci USA 1995; 92: 4342-4246. 266. Hollenbaugh D, Mischel-Petty N, Edwards CP, Simon JC, Denfeld RW, Kiener PA, Aruffo A. Expression of functional CD40 by vascular endothelial cells. J Exp Med 1995; 182: 33-40. 267. Galy AHM, Spits H. CD40 is functionally expressed on human thymic epithelium. J Immunol 1992; 149: 775-782. 268. Banchereau J, Bazan F, Blanchard D, Briere F, Galizzi JP, van Kooten C, Liu YJ, Rousset F, Saeland S. The CD40 antigen and its ligand. Annu Rev Immunol 1994; 12: 881-922. 269. Clark LB, Foy TM, Noelle RJ. CD40 and its ligand. Adv Immunol 1996; 63: 43-78. 270. Foy TM, Aruffo A, Bajorath J, Buhlmann JE, Noelle RJ. Immune regulation by CD40 and its ligand gp39. Annu Rev Immunol 1996; 14: 591-617. 271. Durie FH, Foy TM, Masters SR, Laman JD, Noelle RJ. The role of CD40 in the regulation of humoral and cell-mediated immunity. Immunol Today 1994; 15: 406-411. 272. Campbell KA, Ovendale PJ, Kennedy MK, Fanslow WC, Reed SG, Maliszewski CR. CD40 ligand is required for protective cell-mediated immunity to Leishmania major. Immunity 1996; 4: 283-289. 273. Kamanaka M, Yu P, Yasui T, Yoshida K, Kawabe T, Horii T, Kishimoto T, Kikutani H. Protection role of CD40 in Leishmania major infection at two distinct phases of cell-mediated immunity. Immunity 1996; 4: 275-281. 274. Soong L, Xu J-C, Grewal IS, Kima P, Sun J, Longley Jr BJ, Ruddle NH, McMahon-Pratt D, Flavell RA. Disruption of CD40-CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity 1996; 4: 263-273. 275. Stuber E, Strober W, Neurath M. Blocking the CD40L-CD40 interaction in vivo specifically prevents the priming of T helper 1 cells through the inhibition of interleukin 12 secretion. J Exp Med 1996; 183: 693-698. 276. MacMicking J, Xie Q-w, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol 1997; 15: 323-350. 277. Tian L, Noelle RJ, Lawrence DA. Activated T cells enhance nitric oxide production by murine splenic macrophages through gp39 and LFA-1. Eur J Immunol 1995; 25: 306-309. 278. Stout RD, Suttles J. The many roles of CD40 in cell-mediated inflammatory responses. Immunol Today 1996; 17: 487-492. 279. Larsen CP, Alexander DZ, Hollenbaugh D, Elwood ET, Ritchie SC, Aruffo A, Hendrix R, Pearson TC. CD40-gp39 interactions play a critical role during allograft rejection. Transplantation 1996; 61: 4-9. 280. Jenkins MK. The ups and downs of T cell costimulation. Immunity 1994; 1: 443-446. 281. Roy M, Aruffo A, Ledbetter J, Linsley P, Kehry M, Noelle R. Studies on the interdependence of gp39 and B7 expression and function during antigen-specific immune responses. Eur. J. Immunol. 1995; 1995: 596-603. 282. Ranheim EA, Kipps TJ. Activated T cells induce expression of B7/BB1 on normal or leukemic B cells through a CD40-dependent signal. J Exp Med 1993; 177: 925-935. 283. Kennedy MK, Mohler KM, Shanebeck KD, Baum PR, Picha KS, Otten-Evans CA, Janeway Jr CA, Grabstein KH. Induction of B cell costimulatory function by recombinant murine CD40 ligand. Eur J Immunol 1994; 24: 116-123. 284. Cayabyab M, Philips JH, Lanier LL. CD40 preferentially costimulates activation of CD40+ T lymphocytes. J Immunol 1994; 152: 1523-1531. 285. van Essen D, Kikutani H, Gray D. CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature 1995; 378: 620-623. 286. Opelz G, Sengar DPS, Mickey MR, Terasaki PI. Effect of blood transfusions on subsequent kidney transplants. Transplant Proc 1973; 5: 253-259. 287. Fabre JW, Bishop M, Sen T, McKenzie J, Williams KA, Denton TG, Millard PR, Morris PJ. A study of three protocols of blood transfusion before renal transplantation in the dog. Transplantation 1978; 26: 94-98. 288. van der Linden CJ, Buurman WA, Vegt PA, Greep JM, Jeekel JJ. Effect of blood transfusions on canine renal allograft survival. Transplantation 1982; 33: 400-402. 289. van Es AA, Marquet RL, van Rood JJ, Balner H. Influence of a singe blood transfusion on kidney allograft survival in unrelated rhesus monkeys. Transplantation 1978; 26: 325-330. 290. Wood KJ, Evins J, Morris PJ. suppression of renal allograft rejection in the rat by class I antigens on purified erythrocytes. Transplantation 1985; 39: 56-62. 291. Hibberd AD, Scott LJ. Allogeneic platelets increase the survival of rat renal allografts. Transplantation 1983; 35: 622-624. 292. Lauchart W, Alkins BJ, Davies DAL. Only B lymphocytes induce active enhancement of rat cardiac allografts. Transplantation 1980; 29: 259-261. 293. Nagata M, Ochiai T, Asano T, Gunji Y, Enomoto K, Nakajima K, Uematsu T, Sato H. Role of IA-positive cells in the beneficial effect of donor blood transfusion and induction of suppressor cells in cardiac allotransplantation of rat. Transplantation 1984; 38: 522-526. 294. Jenkins AM, Woodruff MFA. The effect of prior administration of donor strain blood or blood constituents on the survival of cardiac allografts in rats. Transplantation 1971; 12: 57-60. 295. Martin DC, Hewitt CW, Osborne JG, Dowdy SF, Fristoe TL, Russell LA, Cote J. Enhanced kidney graft survival in rats by single or multiple blood transfusion(s) and various blood products. Transplant Proc 1982; 14: 407-409. 296. Superina RA, Wood KJ, Morris PJ. The effect of pretreatment with a single cloned donor class I gene product on cardiac allograft survival in mice. Transplantation 1987; 44: 719-721. 297. Madsen JC, Supeina RA, Wood KJ. Immunological unresponsiveness induced by recipient cells transfected with donor MHC genes. Nature 1988; 332: 161-164. 298. Starzl TE. Heterologous antilymphocyte globulin. N Engl J Med 1968; 279: 700-703. 299. Heyworth MF. Clinical experience with antilymphocyte serum. Immunol Rev 1982; 65: 79-97. 300. Cobbold SP, Adams E, Marshall SE, Davies JD, Waldmann H. Mechanism of peripheral tolerance and suppression induced by monoclonal antibodies to CD4 and CD8. Immunol Rev 1996; 149: 5-33. 301. Kupiec-Weglinski JW, Diamantstein T, Tilney NL. Interleukin 2 receptor-targeted therapy-rationale and applications in organ transplantation. Transplantation 1988; 46: 785-792. 302. Swain SL. T cell subsets and the recognition of MHC class. Immunol Rev 1983; 74: 129-142. 303. Cammarota G, Scheirle A, Takacs B, Doran DM, Knorr R, Bannwarth W, Guardiola J, Sinigaglia F. Identification of a CD4 binding site on the b2 domain of HLA-DR molecules. Nature 1992; 356: 799-801. 304. Vignali DAA, Moreno J, Schiller D, Hammerling GJ. Species-specific binding of CD4 to the b2 domain of major histocompatibility complex class II molecules. J Exp Med 1992; 175: 925-932. 305. Cobbold SP, Jayasuriya A, Nash A, Prospero TD, Waldmann H. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature 1984; 312: 548-551. 306. Cosimi AB, Delmonico FL, Wright KJ, Wee SL, Preffer FI, Jolliffe LK, Colvin RB. Prolonged survival of nonhuman primate renal allograft recipients treated with only anti-CD4 monoclonal antibody. Surgery 1990; 108: 406-413. 307. Wood KJ, Pearson TC, Darby C, Morris PJ. CD4: a potential target molecule for immunosuppressive therapy and tolerance induction. Transplant Rev 1991; 5: 150-164. 308. Madsen JC, Peugh WN, Wood KJ, Morris PJ. The effect of anti-L3T4 monoclonal antibody treatment on first-set rejection of murine cardiac allografts. Transplantation 1987; 44: 849-852. 309. Shizuru JA, Seybel KB, Flavin TF. Induction of donor-specific unresponsiveness to cardiac allografts in rats by pretransplant anti-CD4 monoclonal antibody therapy. Transplantation 1990; 50: 366-373. 310. Shizuru JA, Gregory AK, Chao CTB, Fathman CG. Islet allograft survival after a single course of treatment of recipients with antibody to L3T4. Science 1987; 237: 278-280. 311. Cobbold S, Waldmann H. Skin allograft rejection by L3T4+ and LYT-2+ T cell subsets. Transplantation 1986; 41: 634-639. 312. Lehmann M, Sternkopf F, Metz F, Brock J, Docke WD, Plantikow A, Kuttler B, Hahn HJ, Ringel B, Volk HD. Induction of long-term survival of rat skin allografts by a novel highly efficient anti-CD4 monoclonal antibody. Transplantation 1992; 54: 959-962. 313. Waldmann H. Manipulation of T-cell responses with monoclonal antibodies. Annu Rev Immunol 1989; 7: 407-444. 314. Darby CR, Morris PJ, Wood KJ. Evidence that long-term cardiac allograft survival induced by anti-CD4 monoclonal antibody does not require depletion of CD4+ T cells. Transplantation 1992; 54: 483-90. 315. Chen Z, Cobbold S, Metcalfe S, Waldmann H. Tolerance in the mouse to major histocompatibility complex-mismatched heart allografts, and to rat heart xenografts, using monoclonal antibodies to CD4 and CD8. Eur J Immunol 1992; 22: 805-810. 316. Hynes RO. Integrins: versatility, modulation, and signalling in cell adhesion. Cell 1992; 69: 11. 317. Dustin ML, Springer TA. Lymphocyte function-associated antigen-1 (LFA-1) interaction with intercellular adhesion molecule-1 (ICAM-1) is one of at least three mechanisms for lymphocyte adhesion to cultured endothelial cell. J Cell Biol 1988; 107: 321-331. 318. Dustin ML, Springer TA. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature 1989; 341: 619-624. 319. Bachmann MF, McKall-Faienza K, Schmits R, Bouchard D, Beach J, Speiser DE, Mak TW, Ohashi PS. Distint roles for LFA-1 and CD28 during activation of naive T cells: Adhesion versus Costimulation. Immunity 1997; 7: 549-557. 320. Benjamin RJ, Qin S, Wise MP, Cobbold SP, Waldmann H. Mechanism of monoclonal antibody-facilitated tolerance induction: a possible role for the CD4 (L3T4) and CD11a (LFA-1) molecules in self-nonself discrimination. Eur J Immunol 1988; 18: 1079-1088. 321. Kirkman RL, Barret LV, Gaulton GN, Kelley VE, Ythier A, Strom TB. Administration of an anti-interleukin-2 monoclonal antibody prolongs cardiac allograft survival in mice. J Exp Med 1985; 162: 358-362. 322. Kupiec-Weglinski JW, Diamantstein T, Tilney NL, Strom TB. Therapy with monoclonal antibody to interleukin-2 receptor spares suppressor T cells and prevents or reverses acute allograft rejection. Proc Natl Acad Sci USA 1986; 83: 2624-2627. 323. Lenschow DJ, Zeng Y, Thistlethwaite JR, Montag A, Brady W, Gibson MG, Linsley PS, Bluestone JA. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4Ig. Science 1992; 257: 789-792. 324. Turka LA, Linsley PS, Lin H, Brady W, Leiden JM, Wei R-Q, Gibson ML, Zheng X-G, Myrdal S, Gordon D, Bailey T, Bolling SF, Thompson CB. T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc Natl Acad Sci USA 1992; 89: 11102-11105. 325. Lin H, Bolling SF, Linsley PS, Wei R-Q, Gordon D, Thompson CB, Turka LA. Long-term acceptance of major histocompatibility complex mismatched cardiac allografts induced by CTLA4Ig plus donor-specific transfusion. J. Exp. Med. 1993; 178: 1801-1806. 326. Bolling SF, Lin H, Turka LA. The time course of CTLA4Ig effect on cardiac allograft rejection. J Surg Res 1996; 63: 320-323. 327. Pearson TC, Alexander DZ, Winn KJ, Linsley PS, Lowry RP, Larsen CP. Transplantation tolerance induced by CTLA4-Ig. Transplantation 1994; 57: 1701-1706. 328. Tepper MA, Linsley PS, Tritschler D, Esselstyn JM. Tolerance induction by soluble CTLA4 in a mouse skin transplant model. Transplant Proc 1994; 26: 3151-3154. 329. Larsen CP, Elwood ET, Alexander DZ, Ritchie SC, Hendrix R, Tucker-Burden C, Cho HR, Aruffo A, Hollenbaugh D, Linsley PS, Winn KJ, Pearson TC. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature 1996; 381: 434-438. 330. Sun H, Subbotin V, Chen C, Aitouche A, Valdivia LA, Sayegh MH, Linsley PS, Fung JJ, Starzl TE, Ral AS. Prevention of chronic rejection in mouse aortic allografts by combined treatment with CTLA4-Ig and anti-CD40 Ligand monoclonal antibody. Transplantation 1997; 64: 1838-1843. 331. Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, Hong X, Thomas D, Fechner JH, Knechtel SJ. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA 1997; 94: 8789-8794. 332. Pearson TC, Madsen JC, Larsen CP, Morris PJ, Wood KJ. Induction of transplantation tolerance in adults using donor antigen and anti-CD4 monoclonal antibody. Transplantation 1992; 54: 475-483. 333. Saitovitch D, Morris PJ, Wood KJ. Recipient cells expressing single donor MHC locus products can substitute for donor-specific transfusion in the induction of transplantation tolerance when pretreatment is combined with anti-Cd4 monoclonal antibody. Transplantation 1996; 61: 1532-1538. 334. Bushell AR, Morris PJ, Wood KJ. Tolerance induction by antigen pretreatment and depleting anti-CD4 antibody depends on CD4+ T cell regulation during the induction phase of the response. Eur J Immunol 1995; 25: 2643-2649. 335. Stumbles P, Mason D. Activation of CD4+ T cells in the presence of a nondepleting monoclonal antibody to CD4 induces a Th2-type response in vitro. J Exp Med 1995; 182: 5-13. 336. Buhlmann JE, Foy TM, Aruffo AA, Crassi KM, Ledbetter JA, Green WR, Xu JC, Shultz LD, Roopesian D, Flavell RA, Fast L, Noelle RJ, Durie FH. In the absence of a CD40 signal, B cells are tolerogenic. Immunity 1995; 2: 645-653. 337. Niimi M, Morris PJ, Wood KJ. Small, resting B cells induce prolonged survival of cardiac allografts mismatched for single minor, but not multiple minor, single major, or multiple major and minor histocompatibility antigens. Transplant Proc 1997; 29: 1171. 338. Niimi M, Rawsthorne M-A, Morris PJ, Wood KJ. B7-2+ low density APCs are as effective as B7-2- small resting B cells in inducing specific unresponsiveness to minor histocompatibility (miH) antigen(s) in vivo. In: Ricciardi-Castagnoli P, ed. Dendritic cells in fundamental and clinical immunology. Plenum Publishing Corporation, 1997: 265-269. vol 3). 339. Hollander GA, Castigli E, Kulbacki R, Su M, Burakoff SJ, Ramos J-CG- , Geha RS. Induction of alloantigen-specific tolerance by B cells from CD40-deficient mice. Proc Natl Acad Sci USA 1996; 93: 4994-4998. 340. Parker DC, Greiner DL, Phillips NE, Appel MC, Steele AW, Durie FH, Noelle RJ, Mordes JP, Rossini AA. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc Natl Acad Sci USA 1995; 92: 9560-9564. 341. Markees TG, Phillips NE, Noelle RJ, Shultz LD, Mordes JP, Greiner DL, Rossini AA. Prolongation survival of mouse skin allografts in recipients treated with donor splenocytes and antibody to CD40 ligand. Transplantation 1997; 64: 329-335. 342. Hancock WW, Sayegh MH, Zheng XG, Peach R, Linsley PS, Turka LA. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Natl Acad Sci USA 1996; 93: 13967-13972. 343. Husbands SD, Schonrich G, Arnold B, Chandler PR, Simpson E, Philpott KL, Tomlinson P, O'Reilly L, Cooke A, Mellor AL. Expression of major histocompatibility complex class I antigens at low levels in the thymus induces T cell tolerance via non-deletional mechanism. Eur J Immunol 1992; 22: 2655-2661. 344. Steinman RM, Van Voorhis WC, Spalding DM. Dendritic cells. In: Weir DM, Blackwell C, Herenberg LA, ed. Handbook of experimental immunology. First ed. Blackwell Scientific publications, Oxford, 1986: 1-49. 345. Townsend ARM, Taylor PM, Mellor AL, Askonas BA. Recognition of Db and Kb gene products by influenza-specific cytotoxic T cells. Immunogenetics 1983; 17: 283-294. 346. Defranco AL, Raveche ES, Asofsky R, Paul WE. Frequency of B lymphocytes responsive to anti-immunoglobulin. J Exp Med 1982; 155: 1523-1536. 347. Nonacs R, Humborg C, Tam JP, Steinman RM. Mechanism of mouse spleen dendritic cell function in the generation of influenza-specific cytolytic T lymphocytes. J Exp Med 1992; 176: 519-529. 348. Nabavi N, Freeman GJ, Gault A, Godfrey D, Nadler LM, Glimcher LH. Signalling through the MHC class II cytoplasmic domain is required for antigen presentation and induces B7 expression. Nature 1992; 360: 266-268. 349. Cobbold SP, Martin G, Qin S, Waldmann H. Monoclonal antibodies to promote marrow engraftment and tissue graft tolerance. Nature 1986; 323: 164-6. 350. Metlay JP, Witmer-Pack MD, Agger R, Crowley MT, Lawless D, Steinman RM. The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J Exp Med 1990; 171: 1753-1771. 351. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med 1991; 174: 561-569. 352. Hurme M, Chandler PR, Hetherington CM, Simpson E. Cytotoxic T-cell responses to H-Y: correlation with the rejection of syngeneic male skin grafts. J. Exp. Med 1978; 147: 768-775. 353. Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice: the role of H-2D, H-2K and non H-2 antigens in rejection. Transplantation 1973; 16: 343-350. 354. Superina RA, Peugh WN, Wood KJ, Morris PJ. Assessment of primarily vascularized cardiac allografts in mice. Transplantation 1986; 42: 226-227. 355. Schwartz RH. Costimulation of T lymphocytes: The role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992; 71: 1065-1068. 356. Liu Y, Jones B, Brady W, Janeway CA, Linsley PS. Co-stimulation of murine CD4 T cell growth: cooperation between B7 and heat-stable antigen. Eur J Immunol 1992; 22: 2855-2859. 357. Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science 1990; 248: 1349-1355. 358. Kabelitz D, Pohl T, Pechhold K. Activation-induced cell death (apoptosis) of mature peripheral T lymphocytes. Immunol Today 1993; 14: 338-340. 359. Ucker DS, Hebshi LD, Blomquist JF, Torbett BE. Physiological T-cell death: susceptibility is modulated by activation, aging, and transformation, but the mechanism is constant. Immunol Rev 1994; 142: 273-299. 360. Sena J, Wachtel SS, Murphy G. A comparison of the survival of H-Y incompatible ear, tail and body skin grafts. Transplantation 1976; 21: 412-416. 361. Johnson LL. Apparent lack of H-Y-specific suppressor cells in female mice given male spleen cells intravenously. Transplantation 1990; 49: 152-155. 362. Wachtel SS, Ninnemann JL, Good RA. Skin grafting across H-Y incompatibility after organ culture. Transplantation 1975; 20: 45-48. 363. Hughes-Jones NC, Wickramasinghe SN. Lecture notes on haematology. Oxford: Blackwell scientific publications, 1992 364. Lechler RI, Batchelor JR. Immunogenisity of retransplanted kidney allografts. Effect of inducing chimerism in the first recipient and quantitative studies on immunosuppression of the second recipient. J Exp Med 1982; 156: 1835-41. 365. Roopenian DC, Davis AP, Christianson GJ, Mobraaten LE. The functional basis of minor histocompatibility loci. J Immunol 1993; 151: 4595-4605. 366. Nash JR, Peters M, Bell PRF. Comparative survival of pancreatic islets, heart, kidney, and skin allografts in rats, with and without enhancement. Transplantation 1977; 24: 70-73. 367. Steinmulller D, Wachtel SS. Passenger leukocytes and induction of allograft immunity. Transplant Proc 1980; 12: 100-106. 368. Hart DN, Fabre JW. Demonstration and characterization of Ia positive dendritic cells in the interstitial connective tissue of the rat heart and other tissue, but not brain. J Exp Med 1981; 154: 347-361. 369. Bevan MJ. The major histocompatibility complex determines susceptibility to cytotoxic T cells directed anainst minor histocompatibility antigens. J Exp Med 1975; 142: 1349-1364. 370. Matzinger P, Bevan MJ. Hypothesis: Why do so many lymphocytes respond to major histocompatibility antigens? Cell Immunol 1977; 29: 1-5. 371. Butcher GW, Howard JC. Genetic control of transplant rejection. Transplantation 1982; 34: 161-166. 372. Davis WC, McKenzie IF, Melvold RW. A comparison of skin and heart graft rejection patterns in H-2 mutant mice. Transplantation 1980; 29: 189-192. 373. Hart DN, Fabre JW. Kidney-specific alloantigen system in the rat. Characterization and role in transplantation. J Exp Med 1980; 151: 651-666. 374. Bach FH, Sachs DH. transplantation immunology. N Engl J Med 1987; 318: 489-492. 375. Lagaaij EL, Hennemann IPH, Ruigrok M, de Haan MW, Persijn GG, Termijtelen A, Hendriks GFJ, Weimar W, Claas FHJ, van Rood JJ. Effect of one-HLA-DR-matched and completely HLA-DR-mismatched blood transfusions on survival of heart and kidney allografts. N Engl J Med 1989; 321: 701-705. 376. Lagaaij EL, Ruigrok MB, van Rood JJ, Hendriks GFJ, van der Woude F, Weimar W, van Houwelingen HC, Goulmy E. Blood transfusion induced changes in cell-mediated lympholysis: To immunize or not to immunize. J Immunol 1991; 147: 3348-3352. 377. Weiner HL. Oral tolerance: immune mechanisms and treatment of autoimmune diesase. Immunol Today 1997; 18: 335-343. 378. Fu F, Li Y, Qian S, Lu L, Chambers F, Starzl TE, Fung JJ, Thomson AW. Costimulatory molecule-deficient dendritic cell progenitors (MHC class II+, CD80 dim, CD86-) prolong cardiac allograft survival in nonimmunosuppressed recipients. Transplantation 1996; 62: 659-665. 379. Lu L, McCaslin D, Starzl TE, Thomson AW. Bone marrow-derived dendritic cell progenitors (NLDC145+, MHC class II+, B7-1 dim, B7-2 -) induce alloantigen-specific hyporesponsiveness in murine T lymphocytes. Transplantation 1995; 60: 1539-1545. 380. Rastellini C, Lu L, Ricordi C, Starzl TE, Rao AS, Thomson AW. Granulocytes/macrophage colony-stimulating factor-stimulated hepatic dendritic cell progenitors prolong pancreatic islet allograft survival. Transplantation 1995; 60: 1366-1370. 381. Thomson AW, Lu L, Wan Y, Qian S, Larsen CP, Starzl TE. Identification of donor-derived dendritic cell progenitors in bone marrow of spontaneously tolerant liver allograft recipients. Transplantation 1995; 60: 1555-1559. 382. Austyn JM, Kupiec-Weglinski JW, Hankins DF, Morris PJ. Migration patterns of dendritic cells in the mouse. J Exp Med 1988; 167: 646-651. 383. Arnold B, Schonrich G, Hammerling GJ. Multiple levels of peripheral tolerance. Immunol Today 1993; 14: 12-14. 384. Bell EB, Sparshott SM, Bunce C. CD4+ T-cell memory, CD45R subsets and the persistence of antigen-a unifying concept. Immunol Today 1998; 19: 60-64. 385. Koulova L, Clark EA, Shu G, Dupont B. The CD28 ligand B7/BB1 provides costimulatory signal for alloactivation of CD4+ T cells. J Exp Med 1991; 173: 759-62. 386. Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, Flavell RA. Mice deficient for the CD40 ligand. Immunity 1994; 1: 423-431. 387. Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, Kikutani H. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1994; 1: 167-178. 388. Bluestone JA. New perspectives of CD28-B7-mediated T cell costimulation. Immunity 1995; 2: 555-559. 389. Finkelman FD, Lees A, Morris SC. Antigen presentation by B lymphocytes to CD4+ T cells in vivo: Importance of B lymphocyte and T lymphocyte activation. Sem. Immunol. 1992; 4: 247-252. 390. Parker DC. T cell-dependent B cell activation. Annu Rev Immunol 1993; 11: 331-360. 391. Grewal IS, Flavell RA. A central role of CD40 ligand in the regulation of CD4+ T-cell responses. Immunol Today 1996; 17: 410-414. 392. Cocks BG, de Waal-Malefyt R, Galizzi JP, de Vries JE, Aversa G. IL-13 induces proliferation and differentiation of human B cells activated by the CD40 ligand. Int Immunol 1993; 5: 657-663. 393. Roelen DL, Dover EL, Niimi M, Young NT, Morris PJ, Wood KJ. Semi-allogeneic (F1) versus fully allogeneic blood transfusions: differences in their ability to induce specific immunological unresponsiveness. Eur. J. Immunol. 1996; 26: 1468-1474. 394. Schwartz RH. Acquisition of immunologic self-tolerance. Cell 1992; 57: 1073-1081. 395. Linsley PS, Wallace PM, Johnson J, Gibson MG, Greene JL, Ledbetter JA, Singh C, Tepper MA. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science 1992; 257: 792-795. 396. Castigli E, Alt FW, Davidson L, Bottaro A, Mizoguchi E, Bhan AK, Geha RS. CD40-deficient mice generated by recombination-activating gene-2-deficient blastocyst complementation. Proc Natl Acad Sci USA 1994; 91: 12135-12139. 397. Hasbold J, Johnson-Leger C, Atkins CJ, Clard EA, Klaus GGB. Properties of mouse CD40: Cellular distribution of CD40 and B cell activation by monoclonal anti-mouse CD40 antibodies. Eur J Immunol 1994; 24: 1835-1842. 398. Yang Y, Wilson JM. CD40 ligand-dependent T cell activation: Requirement of B7-CD28 signaling through CD40. Science 1996; 273: 1862-1864. 399. Sprent J, Hurd M, Schaefer M, Heath W. Split tolerance in spleen chimeras. J Immunol 1995; 154: 1198-1206. 400. Kosaka H, Sprent J. Tolerance of CD8+ T cells developing in parent-> F1 chimeras prepared with supralethal irradiation: step-wise induction of tolerance in the intrathymic and extrathymic environments. J Exp Med 1993; 177: 367-378. 401. Field EH, Gao Q, Chen N, Rouse TM. Balancing the immune system for tolerance. Transplantation 1997; 64: 1-7. 402. Fowell D, McKnight AJ, Powrie F, Dyke R, Mason D. Subsets of CD4+ T cells and their roles in the induction and prevention of autoimmunity. Immunol Rev 1991; 123: 37-64. |